

Черепно ключичный дизостоз: Что такое ключично-черепной дизостоз
Что такое ключично-черепной дизостоз
Содержание статьи:
Черепно-ключичный дизостоз — это наследственное заболевание, обусловленное изменениями в гене 6 хромосомы. Оно характеризуется формированием дефектов черепа, а также полным или частичным отсутствием ключиц. Патология внесена в перечень редких болезней. Заболеваемость составляет 1 случай на 1 млн новорожденных детей. Код по МКБ-10 – Q74.0.
Симптомы патологии

Ребенок с черепно-ключичным дизостозом: отмечается выраженное недоразвитие ключиц
Клинические проявления патологии связаны с деформацией черепа и полным или частичным отсутствием ключиц:
- Заметное сужение плечевого пояса, связанное с отсутствием ключиц, надплечья опущены. В плечевых суставах регистрируется патологическое повышение объема движений: ребенок может сомкнуть плечи перед грудиной.
- Выраженная задержка закрытия большого и малых родничков, остаются открытыми швы черепа. Большой родничок часто остается незаращенным в течение всей жизни.
- Нарушение прорезывания молочных, а затем постоянных зубов, изменения локализации корней. Молочные зубы могут сменяться в возрасте 25-30 лет. Наличие сверхкомплектных зубов определяется на рентгенограмме.
- Невысокий рост в сравнении с другими родственниками.
- Изменение формы черепа, которое включает высокий и выдающийся вперед лоб.
- Брахицефалия – «широкая» голова, «большеголовость».
- Глазной гипертелоризм – аномально большое расстояние между парными костями черепа, что выражается широко поставленными глазами.
- Недостаточное развитие костей таза.
Некоторые симптомы черепно-ключичной дисплазии у ребенка могут отсутствовать. Постоянные признаки, наличие которых позволяет заподозрить патологию, включают сужение плечевого пояса, незаращение швов черепа и родничков, а также нарушение прорезывания зубов.
Мутация гена нередко приводит к развитию остеопороза (снижение минеральной плотности костей) и дегенеративно-дистрофической патологии суставов (артроз). Нарушение заращения костных швов и родничков повышает риск развития инфекционного процесса в полости среднего уха.
Причины заболевания
 Ключично-черепной дизостоз развивается вследствие мутации (изменение последовательности нуклеотидов) гена Runx2. Он локализуется в коротком плече 6 хромосомы. Изменение передается по аутосомно-доминантному типу. Это означает, что патология с вероятностью 50% передастся ребенку в случае, если один из родителей имеет мутацию. Частота наследования не зависит от пола ребенка, так как изменение связано с соматической хромосомой. Достоверная причина появления мутации в период внутриутробного развития плода, не связанная с наследственностью, остается невыясненной. Считается, что риск развития любых мутаций генома повышается на фоне воздействия на организм беременной женщины следующих провоцирующих факторов:
Ключично-черепной дизостоз развивается вследствие мутации (изменение последовательности нуклеотидов) гена Runx2. Он локализуется в коротком плече 6 хромосомы. Изменение передается по аутосомно-доминантному типу. Это означает, что патология с вероятностью 50% передастся ребенку в случае, если один из родителей имеет мутацию. Частота наследования не зависит от пола ребенка, так как изменение связано с соматической хромосомой. Достоверная причина появления мутации в период внутриутробного развития плода, не связанная с наследственностью, остается невыясненной. Считается, что риск развития любых мутаций генома повышается на фоне воздействия на организм беременной женщины следующих провоцирующих факторов:
- ионизирующее излучение: радиация, рентген, включая рентгенологические исследования;
- токсические соединения, которые обладают мутагенным действием: ароматические углеводороды, анилиновые красители;
- некоторые медикаментозные средства: цитостатики;
- инфекции: краснуха, токсоплазмоз, хламидиоз.
Исключение воздействия провоцирующих факторов во время беременности позволяет снизить риск развития любых пороков развития.
Методы диагностики

Ключицы при ключично-черепной дисплазии
Заподозрить наличие ключично-черепного дизостоза можно в течение первого года развития ребенка. Во время осмотра обращают внимание на состояние ключиц, сроки зарастания родничков черепа, а также прорезывания зубов. Для верификации патологии используется рентгенологическое исследование черепа, пояса верхних конечностей, рук. При этом выявляют несколько критериев на рентгенограмме, которые необходимы для постановки диагноза:
- Дефекты ключицы – отсутствие наружного (акромиального) конца кости, при этом внутренний конец присутствует. Несколько реже ключица состоит из двух отдельных фрагментов. Редко регистрируется полное отсутствие кости с одной или обеих сторон.
- Наличие сверхкомплектных зубов на рентгенограмме лицевого черепа, нарушение их локализации.
- Изменение формы (деформация) черепа в виде брахицефалии, заметное расширение швов, увеличение родничков в размерах, задержка их зарастания.
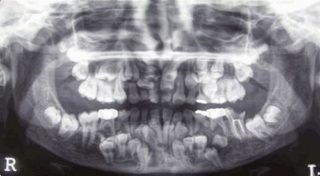
Проблемы зубов
Одновременно назначается диагностика функциональных, органических изменений со стороны других органов и систем. Для этого используются различные методики лабораторного, инструментального исследования. К ним относятся клинический, биохимический анализ крови, общий анализ мочи, компьютерная томография (КТ) или магнитно-резонансная томография (МРТ) различных областей тела, электрокардиография (ЭКГ), электроэнцефалография (ЭЭГ). Количество и направление исследований определяет лечащий врач на основании клинической картины. Качественно проведенная диагностика дает возможность подобрать адекватные терапевтические мероприятия. Это связано с тем, что ключично-черепная дисплазия часто сопровождается нарушениями со стороны различных систем органов.
Ранняя диагностика заболевания проводится еще во время внутриутробного развития плода или непосредственно после рождения ребенка. Она включает использование молекулярно-генетического теста с непосредственным выявлением мутации гена в коротком плече 6 хромосоме.
Если диагноз был установлен на ранних стадиях беременности (до 12 недели) решается вопрос о прерывании по медицинским показаниям.
Методы лечения
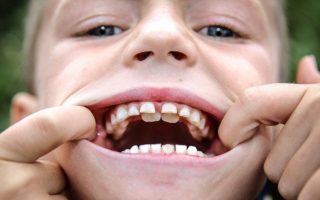
Одним из методов лечения является своевременное удаление сверхкомплектных зубов
Радикальное исправление ключично-черепного дизостоза и восстановление структуры генома 6 хромосомы невозможны. Современная терапия подразумевает применение нескольких направлений восстановительного лечения и пластики костных изменений:
- Своевременное удаление молочных зубов.
- Восстановление зубного ряда (ортодонтическое лечение).
- Удаление сверхкомплектных зубов.
- Костно-пластическая операция, которая назначается при частичном дефекте ключицы и направлена на восстановление анатомической структуры.
Деформация, частичное или полное отсутствие ключицы приводят к сдавливанию плечевого нервного сплетения. Может требоваться выполнение реконструктивного хирургического вмешательства, которое дает возможность избежать дискомфорта и осложнений в будущем. Для правильного формирования скелета пояса верхних конечностей назначаются лечебная гимнастика, массаж, физиотерапия.
Проводится профилактика остеопороза при помощи препаратов, содержащих в своем составе соли кальция, фосфора, витамин Д и его метаболиты. Предотвращение развития дегенеративно-дистрофических процессов в суставах подразумевает назначение лекарств хондропротекторов. Борьба с инфекционными процессами полости носа и среднего уха включает использование антибиотиков широкого спектра действия. Объем и характер терапевтических мероприятий определяются лечащим врачом индивидуально после проведения диагностики.
Прогноз при правильном лечении относительно благоприятный. Дети не отстают в интеллектуальном развитии. Лечебная физкультура дает возможность избежать необратимых изменений скелета в будущем. При выраженных изменениях оформляется инвалидность.

Рентгенограмма черепа показывает открытые швы черепа, большой родничок.

Панорамный снимок челюстей показывает наличие сверхкомплектных зубов.
Ключично-черепной дизостоз (Ключично-черепная дисплазия, синдром Шейтхауэра — Мари — Сентона) — это наследственное заболевание, вызванное мутациями в гене Runx2[1]
Энциклопедичный YouTube
1/1
Просмотров:30 728
✪ Cleidocranial dysplasia (as seen in “Stranger Things”) – causes, symptoms & pathology
Содержание
История
Первый случай отклонений в развитии ключиц был описан М. Мартин в 1765 году. Случай, связанный с отклонениями в развитии ключиц и костей черепа был рассмотрен в 1861 году Шейтхауэром. В 1897 году Мари и Сентон более подробно рассмотрели это заболевание, указали на семейный характер патологии, и дали название dyostosis cleido-cranialis, поскольку соотносили его с дефектами костей черепа и ключиц. Гессе в 1926 году описал отклонения в развитии зубов и челюстей и связал его с ключично-черепным дизостозом.
Симптоматика
Основные симптомы ключично-черепного дизостоза:
- Недоразвитие или отсутствие одной или обеих ключиц. При отсутствии или недоразвитии ключицы плечевой пояс резко сужен, надплечья покаты и опущены. Отмечается избыточная подвижность в плечевых суставах, возможно даже соприкоснуться плечами спереди грудины.
- Задержка закрытия (окостенения) пространства между костями черепа (родничков), могут формироваться дополнительные костные включения. Большой родничок может оставаться открытым в течение всей жизни.
- Нарушения формирования корней, задержка в прорезывании молочных и постоянных зубов. Могут до 25-30-летнего возраста не меняться молочные зубы. Часто встречаются сверхкомплектные зубы.
Также в большинстве случаев отмечено:
- Низкий рост по сравнению с родственниками.
- Брахицефалия.
- Гипертелоризм.
- Высокий и выдающийся вперед лоб.
- Недоразвитие костей таза.
Другие медицинские проблемы включают рецидивы инфекций верхних дыхательных путей; осложнения, рецидивы инфекции ушей; ранний остеопороз и проблемы с суставами; высокую частоту кесарева сечения у женщин; легкую степень моторной задержки у детей в возрасте до пяти лет.
У больных классическим ключично-черепным дизостозом нормальный уровень интеллекта.
Заболевание встречается поровну у мужчин и женщин, и ему подвержены все расы.
Эпидемиология
Распространенность ключично-черепного дизостоза — один на миллион, но это, скорее всего, условно, так как по сравнению с другими скелетными дизостозами относительно мало медицинских осложнений.
Заболевание входит в Перечень редких (орфанных) заболеваний Министерства здравоохранения Российской Федерации[3].
Диагностика
Диагностика ключично-черепного дизостоза основана на клинических симптомах и рентгенологических исследованиях, которые включают изображения черепа, грудной клетки, таза и рук. Главный рентгенологический симптом — дефекты ключиц. Обычно отсутствует наружный (акромиальный) конец ключицы, в то время как внутренний (грудинный) конец присутствует. Но иногда ключица состоит из двух фрагментов. Полное отсутствие ключицы встречается редко.
При молекулярно-генетическом исследовании обнаруживаются мутации гена RUNX2 у 60 % −70 % людей с диагнозом ключично-черепной дизостоз.
Лечение
Заболевание в целом неизлечимо. Но чаще всего необходимо лечение проблем зубов, как наиболее значимых, причем начинать надо с детского возраста:
- Удаление молочных зубов.
- Удаление сверхкомплектных зубов.
- Ортодонтическое лечение.
Неправильность развития ключицы может повлечь за собой сдавление плечевого нервного сплетения, а также общую мышечную слабость верхних конечностей. При резко выраженных явлениях сдавления вполне целесообразно хирургическое вмешательство. При частичном дефекте ключицы возможна костно-пластическая операция — замещение костного дефекта ауто- или аллотрансплантатом. При полном отсутствии ключиц хирургическое лечение нецелесообразно. Консервативная терапия заключается в лечебной гимнастике.
Предотвращение инфекций придаточных пазух носа и среднего уха.
Если плотность костей ниже нормы, то необходимо лечение кальцием и витамином D. Профилактическое лечение при остеопорозе следует начинать в раннем возрасте.
Примечания
Ссылки
 Эта страница в последний раз была отредактирована 15 июля 2020 в 18:18.
Эта страница в последний раз была отредактирована 15 июля 2020 в 18:18.Ключично-черепной дизостоз — Википедия
Материал из Википедии — свободной энциклопедии
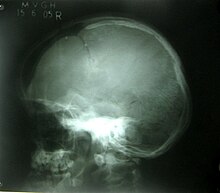 Рентгенограмма черепа показывает открытые швы черепа, большой родничок.
Рентгенограмма черепа показывает открытые швы черепа, большой родничок.  Панорамный снимок челюстей показывает наличие сверхкомплектных зубов.
Панорамный снимок челюстей показывает наличие сверхкомплектных зубов.Ключично-черепной дизостоз (Ключично-черепная дисплазия, синдром Шейтхауэра — Мари — Сентона) — это наследственное заболевание, вызванное мутациями в гене Runx2[1][2], расположенном на коротком плече 6 хромосомы. Ключично-черепной дизостоз передается по аутосомно-доминантному типу. Характеризуется дефектами развития костей черепа, а также полным или частичным отсутствием ключиц.
История
Первый случай отклонений в развитии ключиц был описан М. Мартин в 1765 году. Случай, связанный с отклонениями в развитии ключиц и костей черепа был рассмотрен в 1861 году Шейтхауэром. В 1897 году Мари и Сентон более подробно рассмотрели это заболевание, указали на семейный характер патологии, и дали название dyostosis cleido-cranialis, поскольку соотносили его с дефектами костей черепа и ключиц. Гессе в 1926 году описал отклонения в развитии зубов и челюстей и связал его с ключично-черепным дизостозом.
Симптоматика
Основные симтомы ключично-черепного дизостоза:
- Недоразвитие или отсутствие одной или обеих ключиц. При отсутствии или недоразвитии ключицы плечевой пояс резко сужен, надплечья покаты и опущены. Отмечается избыточная подвижность в плечевых суставах, возможно даже соприкоснуться плечами спереди грудины.
- Задержка закрытия (окостенения) пространства между костями черепа (родничков), могут формироваться дополнительные костные включения. Большой родничок может оставаться открытым в течение всей жизни.
- Нарушения формирования корней, задержка в прорезывании молочных и постоянных зубов. Могут до 25-30-летнего возраста не меняться молочные зубы. Часто встречаются сверхкомплектные зубы.
Также в большинстве случаев отмечено:
- Низкий рост по сравнению с родственниками.
- Брахицефалия.
- Гипертелоризм.
- Высокий и выдающийся вперед лоб.
- Недоразвитие костей таза.
Другие медицинские проблемы включают рецидивы инфекций верхних дыхательных путей; осложнения, рецидивы инфекции ушей; ранний остеопороз и проблемы с суставами; высокую частоту кесарева сечения у женщин; легкую степень моторной задержки у детей в возрасте до пяти лет.
У больных классическим ключично-черепным дизостозом нормальный уровень интеллекта.
Заболевание встречается поровну у мужчин и женщин, и ему подвержены все расы.
Эпидемиология
Распространенность ключично-черепного дизостоза — один на миллион, но это, скорее всего, условно, так как по сравнению с другими скелетными дизостозами относительно мало медицинских осложнений.
Заболевание входит в Перечень редких (орфанных) заболеваний Министерства здравоохранения Российской Федерации[3].
Диагностика
Диагностика ключично-черепного дизостоза основана на клинических симптомах и рентгенологических исследованиях, которые включают изображения черепа, грудной клетки, таза и рук. Главный рентгенологический симптомом — дефекты ключиц. Обычно отсутствует наружный (акромиальный) конец ключицы, в то время как внутренний (грудинный) конец присутвует. Но иногда ключица состоит из двух фрагментов. Полное отсутствие ключицы встречается редко.
При молекулярно-генетическом исследовании обнаруживаются мутации гена RUNX2 у 60 % −70 % людей с диагнозом ключично-черепной дизостоз.
Лечение
Заболевание в целом неизлечимо. Но чаще всего необходимо лечение проблем зубов, как наиболее значимых, причем начинать надо с детского возраста:
- Удаление молочных зубов.
- Удаление сверхкомплектных зубов.
- Ортодонтическое лечение.
Неправильность развития ключицы может повлечь за собой сдавление плечевого нервного сплетения, а также общую мышечную слабость верхних конечностей. При резко выраженных явлениях сдавления вполне целесообразно хирургическое вмешательство. При частичном дефекте ключицы возможна костно-пластическая операция — замещение костного дефекта ауто- или аллотрансплантатом. При полном отсутствии ключиц хирургическое лечение нецелесообразно. Консервативная терапия заключается в лечебной гимнастике.
Предотвращение инфекций придаточных пазух носа и среднего уха.
Если плотность костей ниже нормы, то необходимо лечение кальцием и витамином D. Профилактическое лечение при остеопорозе следует начинать в раннем возрасте.
Примечания
Ссылки
Черепно-ключичный дизостоз: симптомы и лечение патологии
Содержание:
- Симптомы патологии
- Лечение
Черепно-ключичный дизостоз еще называют синдромом Шейтхауэра-Мари-Сентона, или ключично-черепной дисплазией. Это наследственное заболевание, причиной развития которого является мутация одного из генов в 6-й хромосоме. Передается болезнь по аутосомно-доминантному типу. Данная патология характеризуется частичным или полным отсутствием ключицы, а также дефектами в развитии костей черепа.
Это не единственная существующая форма такой патологии, существует еще черепно-лицевой и челюстно-лицевой дизостоз, а также еще ряд форм. Их довольно часто путают, хотя существует ряд отличий.

Ключично-черепной дизостоз
Симптомы патологии
Основными симптомами наличия данной патологии являются:
- отсутствие или недоразвитие обеих или одной из ключиц, из-за чего плечевой пояс сильно сужен, а надплечья опущены. По этой же причине в плечевых суставах заметна чрезмерная подвижность;
- задержка окостенения родничков, при этом в некоторых случаях формируются дополнительные костные включения. В некоторых случаях большой родничок так и не закрывается на протяжении всей жизни человека.;
- присутствие нарушений в формировании корней зубов, а также задержки при прорезывании постоянных и молочных зубов. Иногда у людей с этой патологией молочные зубы не меняются до 30 лет, но при этом отмечается наличие сверхкомплектных зубов.
В большинстве случаев отмечаются также такие симптомы, как брахицефалия, низкий рост (в сравнении с родственниками), гипертелоризм, недоразвитие костей таза, выдающийся вперед и высокий лоб.
У пациентов с классической формой этой патологии обычно нормальный уровень интеллекта, но существует ряд других медицинских проблем. Так, такие люди подвержены рецидивам и осложнениям инфекций ушей, дыхательных путей, у них рано появляются проблемы с суставами и развивается остеопороз, а у детей отмечают легкую степень моторной задержки.
Лечение
Данная болезнь является неизлечимой, поэтому обычно лечение заключается в борьбе с проявлениями этой патологии. Так, в детском возрасте наиболее значимыми считаются проблемы зубов, потому проводится ортодонтическое лечение, удаление сверхкомплектных и молочных зубов.
Проблемы зубов
Также неправильное развитие костей ключицы может привести к общей мышечной слабости рук и сдавливанию плечевого нервного сплетения, в этом случае необходима хирургическая операция. Если же дефект ключицы только частичный, то возможна замена костного дефекта на специальные алло- или аутотрансплантаты. Но при полном отсутствии ключицы хирургическое лечение обычно бессмысленно и приходится обходиться консервативной терапией.
Также важными задачами является профилактика развития остеопороза и предотвращение развития различных инфекций среднего уха и придаточных пазух носа.
Ключично-черепной дизостоз – это… Что такое Ключично-черепной дизостоз?
Рентгенограмма черепа показывает открытые швы черепа, большой родничок. Панорамный снимок челюстей показывает наличие сверхкомплектных зубов.
У пациентов с классической формой этой патологии обычно нормальный уровень интеллекта, но существует ряд других медицинских проблем. Так, такие люди подвержены рецидивам и осложнениям инфекций ушей, дыхательных путей, у них рано появляются проблемы с суставами и развивается остеопороз, а у детей отмечают легкую степень моторной задержки.

Ключично-черепной дизостоз (Ключично-черепная дисплазия, синдром Шейтхауэра — Мари — Сентона) — это наследственное заболевание, вызванное мутациями в гене Runx2[1][2], расположенном на коротком плече 6 хромосомы. Ключично-черепной дизостоз передается по аутосомно-доминантному типу. Характеризуется дефектами развития костей черепа, а также полным или частичным отсутствием ключиц.
История
Первый случай отклонений в развитии ключиц был описан М. Мартин в 1765 году. Случай, связанный с отклонениями в развитии ключиц и костей черепа был рассмотрен в 1861 году Шейтхауэром. В 1897 году Мари и Сентон более подробно рассмотрели это заболевание, указали на семейный характер патологии, и дали название dyostosis cleido-cranialis, поскольку соотносили его с дефектами костей черепа и ключиц. Гессе в 1926 году описал отклонения в развитии зубов и челюстей и связал его с ключично-черепным дизостозом.
Симптоматика
Основные симтомы ключично-черепного дизостоза:
- Недоразвитие или отсутвие одной или обеих ключиц. При отсутствии или недоразвитии ключицы плечевой пояс резко сужен, надплечья покаты и опущены. Отмечается избыточная подвижность в плечевых суставах, возможно даже соприкоснуться плечами спереди грудины.
- Задержка закрытия (окостенения) пространства между костями черепа (родничков), могут формироваться дополнительные костные включения. Большой родничок может оставаться открытым в течение всей жизни.
- Нарушения формирования корней, задержка в прорезывании молочных и постоянных зубов. Могут до 25-30-летнего возраста не меняться молочные зубы. Часто встречаются сверхкомплектные зубы.
Также в большинстве случаев отмечено:
- Низкий рост по сравнению с родственниками.
- Брахицефалия.
- Гипертелоризм.
- Высокий и выдающийся вперед лоб.
- Недоразвитие костей таза.
Другие медицинские проблемы включают рецидивы инфекций верхних дыхательных путей; осложнения, рецидивы инфекции ушей; ранний остеопороз и проблемы с суставами; высокую частоту кесарева сечения у женщин; легкую степень моторной задержки у детей в возрасте до пяти лет.
У больных классическим ключично-черепным дизостозом нормальный уровень интеллекта.
Заболевание встречается поровну у мужчин и женщин, и ему подвержены все расы.
Распространенность ключично-черепного дизостоза – один на миллион, но это, скорее всего, условно, так как по сравнению с другими скелетными дизостозами относительно мало медицинских осложнений.
Диагностика
Диагностика ключично-черепного дизостоза основана на клинических симптомах и рентгенологических исследованиях, которые включают изображения черепа, грудной клетки, таза и рук. Главный рентгенологический симптомом – дефекты ключиц. Обычно отсутствует наружный (акромиальный) конец ключицы, в то время как внутренний (грудинный) конец присутвует. Но иногда ключица состоит из двух фрагментов. Полное отсутвие ключицы встречается редко.
При молекулярно-генетическом исследовании обнаруживаются мутации гена RUNX2 у 60% -70% людей с диагнозом ключично-черепной дизостоз.
Лечение
Заболевание в целом, конечно, неизлечимо. Но чаще всего необходимо лечение проблем зубов, как наиболее значимых, причем начинать надо с детского возраста:
- Удаление молочных зубов.
- Удаление сверхкомплектных зубов.
- Ортодонтическое лечение.
Неправильность развития ключицы может повлечь за собой сдавление плечевого нервного сплетения, а также общую мышечную слабость верхних конечностей. При резко выраженных явлениях сдавления вполне целесообразно хирургическое вмешательство. При частичном дефекте ключицы возможна костно-пластическая операция – замещение костного дефекта ауто- или аллотрансплантатом. При полном отсутствии ключиц хирургическое лечение нецелесообразно. Консервативная терапия заключается в лечебной гимнастике.
Предотвращение инфекций придаточных пазух носа и среднего уха.
Если плотность костей ниже нормы, то необходимо лечение кальцием и витамином D. Профилактическое лечение при остеопорозе следует начинать в раннем возрасте.
Примечания
Ссылки
ДИЗОСТОЗ — Большая Медицинская Энциклопедия
Дизостоз (dysostosis; греч. dys- + osteon кость + osis) — нарушение развития костей, лежащее в основе врожденных наследственных семейных заболеваний костной системы. Чаще всего возникают аномалии развития костей черепа в сочетании с другими симптомами, однако встречаются множественные и генерализованные поражения костей скелета. Термин «Дизостоз» применяют к генерализованным поражениям скелета — хондродистрофии (см.), гаргоилизму (см.), остеогенезу несовершенному (см.) и т. д.
Важнейшие разновидности Дизостозов: ключично-черепной, черепно-лицевой, челюстно-лицевой и челюстно-черепной.
Ключично-черепной Дизостоз
 Рис. 1. Ключично-черепной Дизостоз (синдром Шейтхауэра—Мари—Сентона) у трех сестер. Отсутствие ключиц у двух сестер приводит к полному соприкосновению плеч.
Рис. 1. Ключично-черепной Дизостоз (синдром Шейтхауэра—Мари—Сентона) у трех сестер. Отсутствие ключиц у двух сестер приводит к полному соприкосновению плеч.Ключично-черепной Дизостоз (синдром Шейтхауэра — Мари — Сентона) характеризуется гипоплазией покровных костей черепа в сочетании с полным или частичным недоразвитием одной или обеих ключиц, т. е. нарушением развития так наз. мембранозных костей. Для Д. этого вида характерно незаращение или позднее заращение черепных швов и родничков, брахицефалия (см.) с преобладанием расширения свода черепа в латеральных направлениях, выдающийся лоб, гипоплазия лицевых костей, гл. обр. верхней челюсти, обусловливающая псевдопрогению (кажущееся увеличение нижней челюсти). Нарушение развития челюстей сопровождается запаздыванием прорезывания зубов. Отсутствие ключиц или частичное недоразвитие их с дефектом внутренних, средних или наружных частей ведет к увеличению подвижности плечевого пояса, а при полном отсутствии их — к полному соприкосновению плеч (рис. 1).
Описанные изменения часто сопровождаются деформациями позвоночника, костей верхних и нижних конечностей, стоп, тазовых костей. Аномалия наследуется по рецессивному и доминантному типу, может быть семейной.
При ключично-черепном дизостозе рентгенологически выявляются многочисленные изменения со стороны скелета, однако наиболее характерны изменения ключиц и костей черепа. Дефекты ключиц чаще симметричны и могут быть разных размеров: от небольших до полного отсутствия ключиц. Чаще же всего отсутствует акромиальный конец ключицы. Свободный конец оставшейся части закруглен, покрыт замыкающей костной пластинкой и связан плотным фиброзным тяжем с акромиальным отростком лопатки. По ходу фиброзного тяжа иногда обнаруживаются костные включения.
При рентгенологическом исследовании черепа определяется брахицефалия: мозговой череп увеличен в поперечнике и уменьшен в передне-заднем размере. Основание черепа укорочено в поперечном направлении и несколько удлинено в продольном. Кости свода, особенно лобная, истончены и как бы раздуты, значительно выдаваясь в стороны. Передний родничок остается незаращенным. В местах перекреста швов могут наблюдаться и дополнительные роднички или дополнительные костные включения в самих швах. Кости лицевого черепа малы, верхнечелюстные пазухи недоразвиты. Размеры нижней челюсти не изменены. Обнаруживаются аномалии прикуса, расположения, формы и сроков прорезывания зубов.
При исследовании скелета туловища и конечностей могут быть обнаружены отклонения в развитии ряда костей: уменьшенные размеры лопаток, крестца, костей таза с отсутствием слияния между собой лобковых, седалищных и подвздошных костей и недоразвитием лобкового симфиза; недоразвитие проксимальных отделов бедер с варусной деформацией их; укорочение или отсутствие ногтевых бугристостей у концевых фаланг пальцев кистей и стоп; незаращение дужек позвонков.
При множественном поражении скелета наличие характерных изменений ключиц делает рентгенологический диагноз достоверным.
Черепно-лицевой Дизостоз
 Рис. 2. Однояйцовые близнецы 13 лет с черепно-лицевым дизостозом (синдром Крузона). Характерны широко расставленные глаза, выражено косоглазие, гипоплазия верхней челюсти.
Рис. 2. Однояйцовые близнецы 13 лет с черепно-лицевым дизостозом (синдром Крузона). Характерны широко расставленные глаза, выражено косоглазие, гипоплазия верхней челюсти.Черепно-лицевой Дизостоз (синдром Крузона, гипертелоризм) — недоразвитие костей черепа, мозга и верхней челюсти в сочетании с преждевременным закрытием черепных швов, экзофтальмом (см.), косоглазием (см.), нистагмом (см.), расстройством зрения. Лоб в области переносицы бугрист, глаза широко расставлены (рис. 2), нос своеобразной крючковидной формы («клюв попугая»), гипоплазия верхней челюсти, псевдопрогения; в резко выраженных случаях наблюдается снижение умственного развития. Наследуется по доминантному типу.
Рентгенологически выявляются изменения черепа. На первый план выступает характерная деконфигурация головы и нарушение нормальных соотношений между мозговым и лицевым черепом: первый уменьшен в размерах, имеет почти шаровидную форму, швы заращены, усилены пальцевые вдавления. Кости свода черепа истончены, несколько выпячиваются кнаружи в области переднего родничка. Основание черепа укорочено и углублено, область турецкого седла сужена, глазницы уплощены.
Кости лицевого черепа малы: верхняя челюсть и носовые кости недоразвиты, нижняя челюсть значительно выдается вперед, в силу чего образуется резкий прогиб носа внутрь.
 Рис. 3. Ребенок с челюстно-лицевым дизостозом. Характерны широкие косо расположенные глазные щели, нарушение развития зубов.
Рис. 3. Ребенок с челюстно-лицевым дизостозом. Характерны широкие косо расположенные глазные щели, нарушение развития зубов.Челюстно-лицевой Дизостоз
Челюстно-лицевой Дизостоз (синдром Берри—Франческетти, синдром Франческетти—Цвалена) — гипоплазия гл. обр. нижней челюсти и скуловых костей, макростомия (своеобразное «рыбье» или «птичье» лицо), широкие косо расположенные глазные щели (рис. 3), с вывороченными и скошенными книзу веками и колобомами в наружных отделах, слепые фистулы от углов рта к ушам, языковидное оволосение щек, нарушения развития зубов, деформация ушных раковин, иногда среднего и внутреннего уха с развитием глухоты, устранимой операцией. В противоположность синдромам Крузона и Апера (см. Апера синдром) определяется сильное развитие лобных пазух. Встречается деформация грудной клетки и позвоночника. Наследуется по доминантному типу.
Челюстно-черепной Дизостоз
Челюстно-черепной Дизостоз (синдром Петерс — Хевельса) — гипоплазия верхней челюсти, скуловых дуг, открытый прикус, прогения (выстояние нижней челюсти), укорочение переднего отдела основания черепа. Аномалия наследуется по доминантному типу.
Существуют другие формы черепных Д.: синдромы Гегенхара, Робена, Франсуа и др. Внешний вид больных с различными формами Д. характерен. Д. сохраняется всю жизнь, не поддается оперативной коррекции, почти не требует дифференциальной диагностики с другими заболеваниями. В сомнительных случаях важным диагностическим методом является рентгенологическое исследование.
Различают так наз. неполные типы перечисленных Д., когда имеют место не все характеризующие их симптомы. Отдельные признаки могут комбинироваться в различных сочетаниях, составляя как бы промежуточные типы Д.
Прогноз для жизни благоприятный.
Библиография: Алексеев В. А. Случай черепно-ключичного дизостоза, Вестн, рентгенол, и радиол., № 3, с. 80, 1974; Косинская Н. С. Нарушения развития костно-суставного аппарата, Л., 1966; Кручинский Г. В. Редкие врожденные синдромы лица и челюстей (в границах первой и второй жаберных дуг), Минск, 1974, библиогр.; Рейнберг С. А. Рентгенодиагностика заболеваний костей и суставов, кн. 1—2, М., 1964; Ромоданов А. П. и Лягценко Д. С. Черепно-лицевой Дизостоз, Журн, невропат, и психиат., т. 72, № 10, с. 1487, 1972, библиогр.; Fleischer-Peters A. Kiefermissbildungen bei Dysostose-Syndromen des Schadels, Dtsch, zahnarztl. Z., Bd 24, S. 932, 1969; Humangenetik, hrsg. v. P. E. Becker, Bd 2, S. 489, Stuttgart, 1964, Bibliogr.; Hylton R. P. a. Albright J. E. Cleidocranial dysostosis, J. oral Surg., v. 28, p. 682, 1970; Tessier P. The definite plastic surgical treatment of the severe facial deformities of craniofacial dysostosis, Plast. reconst. Surg., v. 48, p. 419, 1971.
T. П. Виноградова; И. Г. Лагунова (рент.).
Ключично-черепной дизостоз — Википедия
Материал из Википедии — свободной энциклопедии
Рентгенограмма черепа показывает открытые швы черепа, большой родничок. Панорамный снимок челюстей показывает наличие сверхкомплектных зубов.Ключично-черепной дизостоз (Ключично-черепная дисплазия, синдром Шейтхауэра — Мари — Сентона) — это наследственное заболевание, вызванное мутациями в гене Runx2[1][2], расположенном на коротком плече 6 хромосомы. Ключично-черепной дизостоз передается по аутосомно-доминантному типу. Характеризуется дефектами развития костей черепа, а также полным или частичным отсутствием ключиц.
История
Первый случай отклонений в развитии ключиц был описан М. Мартин в 1765 году. Случай, связанный с отклонениями в развитии ключиц и костей черепа был рассмотрен в 1861 году Шейтхауэром. В 1897 году Мари и Сентон более подробно рассмотрели это заболевание, указали на семейный характер патологии, и дали название dyostosis cleido-cranialis, поскольку соотносили его с дефектами костей черепа и ключиц. Гессе в 1926 году описал отклонения в развитии зубов и челюстей и связал его с ключично-черепным дизостозом.
Симптоматика
Основные симтомы ключично-черепного дизостоза:
- Недоразвитие или отсутствие одной или обеих ключиц. При отсутствии или недоразвитии ключицы плечевой пояс резко сужен, надплечья покаты и опущены. Отмечается избыточная подвижность в плечевых суставах, возможно даже соприкоснуться плечами спереди грудины.
- Задержка закрытия (окостенения) пространства между костями черепа (родничков), могут формироваться дополнительные костные включения. Большой родничок может оставаться открытым в течение всей жизни.
- Нарушения формирования корней, задержка в прорезывании молочных и постоянных зубов. Могут до 25-30-летнего возраста не меняться молочные зубы. Часто встречаются сверхкомплектные зубы.
Также в большинстве случаев отмечено:
- Низкий рост по сравнению с родственниками.
- Брахицефалия.
- Гипертелоризм.
- Высокий и выдающийся вперед лоб.
- Недоразвитие костей таза.
Другие медицинские проблемы включают рецидивы инфекций верхних дыхательных путей; осложнения, рецидивы инфекции ушей; ранний остеопороз и проблемы с суставами; высокую частоту кесарева сечения у женщин; легкую степень моторной задержки у детей в возрасте до пяти лет.
У больных классическим ключично-черепным дизостозом нормальный уровень интеллекта.
Заболевание встречается поровну у мужчин и женщин, и ему подвержены все расы.
Эпидемиология
Распространенность ключично-черепного дизостоза — один на миллион, но это, скорее всего, условно, так как по сравнению с другими скелетными дизостозами относительно мало медицинских осложнений.
Заболевание входит в Перечень редких (орфанных) заболеваний Министерства здравоохранения Российской Федерации[3].
Диагностика
Диагностика ключично-черепного дизостоза основана на клинических симптомах и рентгенологических исследованиях, которые включают изображения черепа, грудной клетки, таза и рук. Главный рентгенологический симптомом — дефекты ключиц. Обычно отсутствует наружный (акромиальный) конец ключицы, в то время как внутренний (грудинный) конец присутвует. Но иногда ключица состоит из двух фрагментов. Полное отсутствие ключицы встречается редко.
При молекулярно-генетическом исследовании обнаруживаются мутации гена RUNX2 у 60 % −70 % людей с диагнозом ключично-черепной дизостоз.
Лечение
Заболевание в целом неизлечимо. Но чаще всего необходимо лечение проблем зубов, как наиболее значимых, причем начинать надо с детского возраста:
- Удаление молочных зубов.
- Удаление сверхкомплектных зубов.
- Ортодонтическое лечение.
Неправильность развития ключицы может повлечь за собой сдавление плечевого нервного сплетения, а также общую мышечную слабость верхних конечностей. При резко выраженных явлениях сдавления вполне целесообразно хирургическое вмешательство. При частичном дефекте ключицы возможна костно-пластическая операция — замещение костного дефекта ауто- или аллотрансплантатом. При полном отсутствии ключиц хирургическое лечение нецелесообразно. Консервативная терапия заключается в лечебной гимнастике.
Предотвращение инфекций придаточных пазух носа и среднего уха.
Если плотность костей ниже нормы, то необходимо лечение кальцием и витамином D. Профилактическое лечение при остеопорозе следует начинать в раннем возрасте.
Примечания
Ссылки
Клеидокраниальный дизостоз – это скелетная дисплазия, наследуемая по аутосомно-доминантному типу, которая может приводить к таким осложнениям, как сколиоз и кифоз, одновременно с различными ортопедическими поражениями. Поскольку сопутствующие деформации позвоночника имеют прогрессирующий характер, может потребоваться хирургическое лечение. В дополнение к другим ортопедическим проблемам следует учитывать возможные сопутствующие осложнения, такие как атлантоаксиальный подвывих, миелопатия, сирингомиелия, врожденные деформации позвоночника, спондилез и спондилолистез при планировании лечения сколиоза и кифоза.Увеличение продолжительности использования благоприятных для роста систем (ростовых стержней) у пациентов, таких как у нас, с ранним появлением симптомов и выполнением задней инструментария и сращения после завершения роста позвоночника даст успешные результаты без осложнений в средней и длинной части. срок. Дальнейшие многоцентровые исследования с более всесторонними оценками необходимы, чтобы найти решения проблем позвоночника, связанных с этой редкой скелетной дисплазией.
1. Введение
Клеидокраниальный дизостоз – это скелетная дисплазия, наследуемая по аутосомно-доминантному типу и характеризующаяся внутримембранозным образованием кости.Это вызывает нарушения в ключице, черепной коробке и тазу. Это расстройство было впервые описано Мари и Сэйнтоном в 1898 году [1, 2] и также известно как кледокраниальная дисплазия, синдром Шойтауэра-Мари-Сэйнтона, мутационный дизостоз, остеодентальная дисплазия, генерализованный дизостоз, тазовая дисклеточная дисфункция тазовых костей [3], дисблазия таза и шейки матки [3] ].
Клеидокраниальный дизостоз – это состояние, унаследованное аутосомно-доминантным способом, при котором у 1/3 пациентов проявляются спонтанные мутации, а у 2/3 – семейные изменения [4].Ответственный ген RUNX2 (связанный с Runt фактор транскрипции 2) представляет собой клонированный ген, расположенный на коротком плече хромосомы 6 (6p21) [5–8]. RUNX2 активирует дифференцировку остеобластов как специфичный для остеобластов фактор транскрипции и регулятор дифференцировки остеобластов [6, 9]. Он также контролирует дифференцировку клеток-предшественников в остеобластах. Клетки секретируют костный матрикс и, таким образом, образуют кость. Кроме того, RUNX2 играет ключевую роль в регуляции дифференцировки хондроцитов во время формирования эндохондральной кости.Этот новый «главный ген» может объяснить основные механизмы формирования кости в дополнение к патобиологии клеидокраниального дизостоза [10].
Характерные признаки клидокраниального дизостоза включают гипоплазию или отсутствие ключицы, брахицефальный череп, гипоплазию в середине лица, отсроченное закрытие родничков и небольшую или умеренную короткую фигуру. Хотя наиболее важные аномалии наблюдаются в реформациях костей посредством внутримембранозного окостенения в ключице, черепе и тазу, рост эндохондральной кости также слегка нарушен и вызывает легкую форму карликовости [2, 4].Задержка прорезывания постоянных зубов и наличие нештатных зубов является значительной причиной заболеваемости [10], что, в свою очередь, требует многочисленных хирургических операций и длительного лечения зубов [1]. В результате неадекватного окостенения контуров зародышевой дуги позвоночника могут развиться деформации позвоночника, такие как расщелина позвоночника, сколиоз, кифоз / кифосколиоз, спондилолиз, спондилолистез, гемивертебра, заднее заклинивание позвонков, могут развиться эти шейные ребра, и могут развиться эти шейные ребра, и могут появиться шейные ребра, и могут появиться шейные ребра, и могут появиться шейные ребра, а также могут появиться шейные ребра, и могут появиться шейные ребра; наблюдается вместе с отсутствием задней грудной позвоночной дуги или сирингомиелией [1, 4, 10, 11].Клеидокраниальный дизостоз имеет предполагаемую распространенность 1/1 000 000 (Таблица 1). Однако, из-за отсутствия диагноза, его распространенность, по оценкам, выше, без различий между полами и этническими группами [1].
131414 6212 2 9203 Рост20 900 900 9 9 9008
Чтобы лучше понять эту редкую скелетную дисплазию и сопровождающие ее деформации позвоночника, мы представляем в этом исследовании результаты лечения у двух пациентов с клеидокраниальным дизостозом и обзор литературы. 2. История болезниМы выполнили слияние задних конечностей и инструментарий из-за прогрессирующего сколиоза у двух пациенток-подростков, у которых диагностировали клеидокраниальный дизостоз после генетического скрининга. Оба пациента имели положительную семейную историю этого состояния. Помимо деформаций позвоночника, мы тщательно изучили сопутствующие ортопедические и стоматологические проблемы пациентов. Средний возраст пациентов составлял 12 (от 11 до 13) лет на момент операции, а средний период наблюдения составил 11 (от 6 до 16) лет.Клинические и рентгенологические результаты были оценены ретроспективно (Таблица 2).
В первом случае (28-летняя женщина) у пациента были типичные фенотипические признаки дисидозов клеидокраниальных (невысокий рост, открытый передняя родничок, типичный внешний вид лица, широкий и выпуклый лоб и проблемы с зубами), двусторонний псевдоартроз ключицы, слегка расширенный лобковый симфиз, маленькие крылья подвздошной кости, двусторонняя одышка шейки бедра и тазобедренная кость, двусторонняя половая щель в нижней конечности , прогрессирующий сколиоз и положительная семейная история (у ее отца и бабушки) на презентации.Двусторонняя остеотомия проксимального отдела большеберцовой кости и вариация с использованием внешних фиксаторов были выполнены для лечения деформации гена valgum (в возрасте 11 лет). Коррекция прогрессирующей деформации сколиоза и слияния была достигнута с помощью винтов с задней ножкой и фиксации крюка (13 лет). Никаких осложнений в течение обычного периода наблюдения в 16 лет не наблюдалось (рисунки 1 и 2). Клеидокраниальный дизостоз – Википедия переиздано // WIKI 2 Клеидокраниальный дизостоз ( CCD ), также называемый Клеидокраниальная дисплазия , является врожденным дефектом, который в основном поражает кости и зубы. [1] Ключицы, как правило, плохо развиты или отсутствуют, что позволяет сближать плечи. [1] Передняя часть черепа часто не закрывается до позднего времени, а пораженные лица часто короче среднего. [1] Другие симптомы могут включать выпуклый лоб, широко расставленные глаза, ненормальные зубы и плоский нос. [1] Симптомы у разных людей разные; однако интеллект, как правило, не подвержен влиянию. [1] Условие либо наследуется от родителей человека, либо возникает как новая мутация. [1] Он наследуется по аутосомно-доминантному типу. [1] Это связано с дефектом в гене RUNX2, который участвует в формировании кости. [1] Диагноз подозревается на основании симптомов и рентгеновских лучей с подтверждением генетическим тестированием. [4] Другие состояния, которые могут вызывать сходные симптомы, включают дисплазию нижней челюсти, пикнодизостоз, несовершенный остеогенез и синдром Хайду-Чейни. [5] Лечение включает вспомогательные меры, такие как устройство для защиты черепа и уход за зубами. [5] Хирургия может быть выполнена, чтобы исправить некоторые аномалии кости. [4] Ожидаемая продолжительность жизни в целом нормальная. [3] Поражает около одного на миллион человек. [1] Мужчины и женщины поражаются одинаково часто. [5] Современное описание даты состояния по крайней мере до 1896 года. [6] Термин от cleido означает ключица, черепная часть от греческого κρανιὀς означает череп и дизостоз означает формирование аномальной кости , [7] Энциклопедия YouTube
СодержаниеПризнаки и симптомыКлеидокраниальный дизостоз – это общее состояние скелета [8] , названное так из-за деформаций ключицы (ключицы) и черепа, которые часто возникают у людей с ним. Люди с этим заболеванием обычно имеют безболезненный отек в области ключиц в возрасте 2–3 лет. [9] Общие черты:
Другими особенностями являются: париетальный босс, базилярный [необходимо устранение неоднозначности ] инвагинация (атлантоаксиальная деформация), постоянный метопический шов, аномальные структуры уха с потерей слуха, нештатные ребра, гемивертебра со спондилезом, малые и высокие лопатки, гипоплазия заболела , отсутствие лобковой кости, короткие / отсутствующие малоберцовые кости, короткие / отсутствующие лучевые кости, гипопластические терминальные фаланги. [16] ГенетикаОбычно это аутосомно-доминантный, но в некоторых случаях причина неизвестна. [17] Это происходит из-за гаплонедостаточности, вызванной мутациями в гене CBFA1 (также называемом Runx2), расположенном на коротком плече хромосомы 6, который кодирует фактор транскрипции, необходимый для дифференцировки остеобластов. [10] Это приводит к отсроченной окостенению срединных структур тела, особенно перепончатой кости. В новой статье сообщается, что причина ПЗС, как полагают, связана с дефектом гена CBFA1 (активность связующего фактора ядра 1) на коротком плече хромосомы 6p21.CBFA1 жизненно важен для дифференцировки стволовых клеток в остеобласты, поэтому любой дефект этого гена будет вызывать дефекты образования мембранных и эндохондральных костей. [18] ДиагнозРазличные признаки дизостоза являются значительными. Радиологическая визуализация помогает подтвердить диагноз. Во время беременности (беременности) размер ключицы может быть рассчитан с использованием доступных номограмм. Иногда в черепе можно наблюдать кости червя. [19] Диагностика нарушения спектра ПЗС устанавливается у индивидуума с типичными клиническими и рентгенологическими данными и / или путем идентификации гетерозиготного патогенного варианта в RUNX2 (CBFA1). [20] ЛечениеОколо 5 лет хирургическая коррекция может быть необходима, чтобы предотвратить любое ухудшение деформации. [9] Если у матери дисплазия, может потребоваться кесарево сечение. Черепно-лицевая хирургия может быть необходима для исправления дефектов черепа. [19] Coxa vara лечат с помощью корректирующих остеотомий бедра. Если имеется раздражение плечевого сплетения с болью и онемением, может быть выполнено удаление фрагментов ключицы для его декомпрессии. [10] В случае открытого родничка ортопед может посоветовать соответствующий головной убор для защиты от травм. ПрогнозВ нескольких исследованиях сообщается, что ожидаемая продолжительность жизни у людей с ПЗС нормальна. [3] [21] [22] ЭпидемиологияКлеидокраниальный дизостоз поражает около одного человека на миллион. [1] Известные случаиВ 1987 году молодая девушка по имени Джессика МакКлюр упала с узкой скважины в доме своей семьи в Техасе.Рон Шорт, подрядчик по кровельным работам, который родился без ключицы из-за клецокраниального дизостоза и, таким образом, мог сломать плечи, чтобы работать в тесных углах, прибыл на участок и предложил спуститься по шахте. Спасатели не использовали его, [23] [24] , хотя МакКлюр был успешно извлечен из скважины. Детский актер Гатен Матараццо родился с кледокраниальной дисплазией, которая включена в сюжетную линию его персонажа Дастина Хендерсона на Stranger Things . б с д е F г ч я J к л м н o “Клеидокраниальная дисплазия”. Старр, Майкл (11 июня 2020 года). «Встреча нового„Дум Патруль“со-звезда Эбигейл Шапиро». New York Post . Получено 15 июня 2020 г. Внешние ссылки Последний раз эта страница редактировалась 24 июля 2020 года, в 01:43.
, Клеидокраниальный дизостоз – Инфогалактика: планетарное ядро знаний Клеидокраниальный дизостоз , также называемый Клеидокраниальная дисплазия или мутационный дизостоз , является наследственным врожденным заболеванием, при котором наблюдается отсроченная окостенение срединных структур. ЭтиопатогенезОбычно это аутосомно-доминантный, но в некоторых случаях причина неизвестна. [1] Это происходит из-за гаплонедостаточности, вызванной мутациями в гене CBFA1 (также называемом Runx2), расположенном на коротком плече хромосомы 6, который кодирует фактор транскрипции, необходимый для дифференцировки остеобластов. [2] Это приводит к отсроченной окостенению срединных структур тела, особенно перепончатой кости. В новой статье сообщается, что этиология ПЗС, как полагают, обусловлена дефектом гена CBFA1 (активностью связующего фактора 1) на коротком плече хромосомы 6p21. CBFA1 жизненно важен для дифференцировки стволовых клеток в остеобласты, поэтому любой дефект этого гена будет вызывать дефекты образования мембранных и эндохондральных костей. [3] Клинические особенностиКлеидокраниальный дизостоз – это общее состояние скелета [4] , названное так по деформации ключицы (cleido-) и черепной коробки, которые часто возникают у людей с ней. Люди с этим заболеванием обычно имеют безболезненный отек в области ключиц в возрасте 2–3 лет. [5] Общими признаками являются:
Другими особенностями являются: париетальное выпячивание, базилярная инвагинация (атлантоаксиальное наложение), стойкий метопический шов, аномальные ушные структуры с потерей слуха, неумеренные ребра, гемивертебра со спондилезом, малые и высокие лопатки, гипоплазия подвздошных костей, отсутствие лобковой кости, короткие / отсутствующие малоберцовые кости, короткие / отсутствующие лучевые кости, гипопластические терминальные фаланги. [11] Диагноз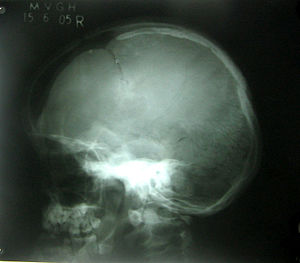 Клинически, различные признаки дизостоза являются значительными. Радиологическая визуализация помогает подтвердить диагноз. Во время беременности (беременности) размер ключицы может быть рассчитан с использованием доступных номограмм. Иногда в черепе можно наблюдать кости червя. [12] ЛечениеОколо 5 лет хирургическая коррекция может быть необходима, чтобы предотвратить любое ухудшение деформации. [5] Если у матери дисплазия, необходима кесарево сечение.Черепно-лицевая хирургия может быть необходима для исправления дефектов черепа. [12] Кокса вара лечится с помощью корректирующих остеотомий бедра. Если имеется раздражение плечевого сплетения с болью и онемением, может быть выполнено удаление фрагментов ключицы для его декомпрессии. [2] В случае открытого родничка ортопед может посоветовать соответствующий головной убор для защиты от травм. ЭпидемиологияЗаболеваемость оценивается в 1: 200 000. [12] Известные случаиУ комика Эммета Ферроу нет ключицы и он использует полученную дополнительную подвижность плеч в рутинных комедиях. На помощь Джессике МакКлюр, Рон Шорт, мускулистый мужчина (кровельный подрядчик), который родился без ключицы из-за клецокраниального дизостоза и поэтому мог сломать плечи, чтобы работать в стесненных углах, прибыл на место и предложил спуститься вниз. вал; они приняли его предложение, хотя и не использовали его. [13] [14] Рекомендации
Внешние ссылки,Disostosis cleidocraneal – Wikipedia, la enciclopedia libre La disostosis cleidocraneal , también llamada dislasia cleidocraneal , es una enfermedmed rara de origen genético que se caccteriza porразнижение, аномалию que afectan al desarrolne de la huesos al al es alla é la uni v al al al en un esna de la es – alla ç l? Al en un esna unos – unicos de lla esca – no losos – alla é l? Al l uni uni un возвращаемость де лос диентес [1] Описание основных событий 1765 года. Происходящее из первых рук в истории Марии Сентона 1897 года. [3] [4] Etiología y herencia [editar]Середина срока годности и законности, в том числе и исходная информация о вероятности возникновения 50% вероятности появления фактических данных.Причинно-следственная связь между генами RUNX2 и человеческими факторами (6p21). [1] Сводная информация о протекционистской деятельности, в том числе о фактической транскрипции, и ее формальностях, касающихся различий в образовании и остеобластах. Он обнаруживает различные мутации, вызывающие провокацию, провокацию, поддержку, защиту, отсутствие защиты, отсутствие каких-либо общих причин, обусловливающих первичную ситуацию в мире, в том числе и по закону, действующим в целом, в высшей степени защищен от вирусов, в целом SE Pierde. [5]  La maloclusión es uno de los síntomas ma frecuentes de la disostosis cleidocraneal La maloclusión es uno de los síntomas ma frecuentes de la disostosis cleidocraneal Клинические и другие переменные, в том числе индивидуальные, стандартные, мутационные. Общие сведения о наблюдаемых событиях, которые можно наблюдать, составляют всего 1,50 метра в месяц и ниже. Лас родничок постоянный, постоянный, нормальный, в том числе и настойчивый, производящий аббревиатуру во фронтальном и темном отделении, нормальный (гипертелоризм) и эсарольский не полностью esfenoidal. [5] [2] Лос-анджелесская школа, посвященная вопросам, связанным с фальта-де-десарролло-де-лас-клавикулас, которая является приблизительной и близкой к общему мнению. Сыграть в экстремальные ситуации в общем, нормальном, продольном и продольном движении. В общем, устный, изменчивый, аномальный, дефектный, неправильный, отклоняющийся от нормы, имплантантные и ретрассо, де-ла-де-де-ла-де-ла-де-ла-де-ла-втаранс, округ Колумбия Del Esmalte Dental. [2] Diagnostico [editar]Essenible реализует диагностику пренатально, с предварительным отбором, превентивным, в том числе знакомым конкретным образом. [2] Дифференциальный диагноз эссенциальной конъюнктуры, в том числе синдром Крейна-Хейса, пикодизостоз, синдром Юниса-Варона и гипофосфатазии. La picnodisostosis совпадают en las malformaciones del cráneo, estatura baja y alteraciones dentales, per se se asocia fragilidad de los huesos and está provocada por una дефицит де-катепсина К. [6] Tratamiento [editar]В общем и целом, в связи с этим, как правило, так и есть. [2] Отсутствует действующее медицинское лечение, медицинское лечение и реабилитация, лечение и ортопедические заболевания. Это может быть связано с тем, что у вас есть все, что вам нужно. [3] Si las fontanelas перманентные аберриты, в том числе и настоящие, травматические, внутрижелудочные и травматические симптомы.Изучение прав человека и ортодонтии, в том числе важного, важного для всей страны. Основополагающее значение для всех поколений составляет 50% вероятности передачи и передачи данных о происшествии. [3] Referencias [editar], | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||