

Болезнь рихтера что это такое – РИХТЕРА СИНДРОМ это что такое РИХТЕРА СИНДРОМ: определение — Психология.НЭС
Синдром Рихтера или трансформация – Рак
Синдром Рихтера (РС), также известный как трансформация Рихтера, относится к превращению одного конкретного типа рака крови в другой, более агрессивный тип.
РС относится к развитию высококачественная неходжкинская лимфома в человеке, который имеет хронический лимфолейкоз (ХЛЛ) / малая лимфоцитарная лимфома (ХЛЛ) , Известно также, что встречаются и другие варианты RS, такие как трансформация в лимфому Ходжкина. Объяснение этих терминов и их значение следующим образом.
обзор
РС развивается у кого-то, у кого уже есть рак белых кровяных клеток. Этот первый рак имеет два разных названия, в зависимости от того, где в организме обнаружен рак: он называется CLL, если рак в основном обнаруживается в крови и костном мозге, или SLL, если обнаруживается в основном в лимфатических узлах.
CLL используется для охвата обеих сущностей, о чем пойдет речь в этой статье.
Не у всех с ХЛЛ развивается синдром Рихтера
Развитие РС у людей с ХЛЛ относительно редко. Оценки, опубликованные в 2016 году, показывают, что трансформация Рихтера происходит только у 5 процентов пациентов с ХЛЛ. Другие источники приводят диапазон от 2 до 10 процентов. Если RS случается с вами, очень необычно, что это произойдет в то же время, когда диагностируется CLL. Люди, у которых развивается РС от ХЛЛ, обычно делают это через несколько лет после диагноза ХЛЛ.
Новый рак обычно ведет себя агрессивно
Новый рак возникает, когда у человека с ХЛЛ развивается так называемая трансформация, чаще всего с неходжкинской лимфомой высокого уровня (НХЛ). «Высокий уровень» означает, что рак имеет тенденцию расти быстрее и быть более агрессивным. Лимфома – это рак лейкоцитов лимфоцитов.
Согласно одному исследованию, около 90 процентов превращений из CLL в тип НХЛ, называемый диффузной крупной B-клеточной лимфомой (DLBCL), в то время как около 10 процентов превращаются в лимфому Ходжкина. В этом случае его на самом деле называют «вариант Ходжкина-синдрома Рихтера (HvRS)», и неясно, отличается ли прогноз от лимфомы Ходжкина. Другие преобразования из CLL также возможны.
Почему это называется синдром Рихтера?
Мужчина по имени Морис Н. Рихтер впервые описал синдром в 1928 году. Он написал о 46-летнем судоходном клерке, который был помещен в больницу и у него постепенно снижалось течение, ведущее к смерти. В ходе анализа вскрытия он определил, что ранее было одно злокачественное новообразование, но из него, по-видимому, возникло новое злокачественное новообразование, которое быстрее росло, покушалось и разрушило ткань, которая была старой ХЛЛ.
Он предположил, что ХЛЛ существовал гораздо дольше, чем кто-либо знал об этом пациенте, также писал о двух раках или поражениях, заявляя: «Возможно, развитие одного из поражений зависело от существования другого. «.
Характеристики
У людей с РС развивается агрессивное заболевание с быстро увеличивающимися лимфатическими узлами, расширением селезенки и печени и повышенными уровнями маркера в крови, известного как сывороточная лактатдегидрогеназа, или ЛДГ.
Процент выживаемости
Как и во всех лимфомах, статистику выживания трудно интерпретировать. Индивидуальные пациенты различаются по общему состоянию здоровья и силе до постановки диагноза. Кроме того, даже два вида рака с одинаковым могут вести себя по-разному у разных людей. С РС, однако, новый рак более агрессивен. Сообщалось, что у некоторых людей с РС выживаемость составляла в среднем менее 10 месяцев после постановки диагноза. Тем не менее, некоторые исследования показали 17-месячную среднюю выживаемость, а другие люди с РС могут жить дольше; Трансплантация стволовых клеток может дать шанс для длительного выживания.
Признаки и симптомы
Если ваш CLL трансформировался в DLBCL, вы заметите явное ухудшение ваших симптомов. Характеристики РС включают быстрый рост опухоли с или без вовлечения экстранодальных органов, то есть новообразования могут быть ограничены лимфатическими узлами, или рак может включать органы, отличные от лимфатических узлов, такие как селезенка и печень.
Вы можете испытать:
- Быстро увеличивающиеся лимфатические узлы
- Дискомфорт в животе, связанный с увеличенной селезенкой и печенью, называемой
Болезнь Рейтера: симптомы и лечение, фото
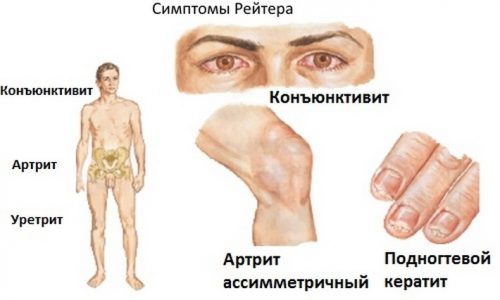
Одним из осложнений хронического хламидиоза выступает болезнь Рейтера, что представляет собой комплексное поражение органов мочеполовой системы, суставов и слизистой оболочки органов зрения. Обычно у человека одновременно или последовательно развивается уретрит, полиартрит и конъюнктивит.
Главной причиной развития патологии выступает аутоиммунный процесс, что спровоцирован микроорганизмами Chlamydia trachomatis, в результате которого нарушается иммунная система человека. Обычно болезнь Рейтера у женщин наблюдается в двадцать раз реже, чем у мужчин, что обусловлено множественными половыми связями последних. Чаще всего патология развивается у военнослужащих и гомосексуалистов.
Характеристика болезни
Болезнь Рейтера – это заболевание ревматического характера, что сочетает в себе одновременное или поочередное поражение органов мочеполовой системы, суставов и глаз. Обычно заболевание развивается в результате воздействия на организм бактерий Chlamydia trachomatis, которые попадают в него половым или контактно-бытовым путем. Хламидии длительное время могут паразитировать в клетках инфицированного человека. В некоторых случаях развитие патологии провоцируют такие инфекции, как сальмонеллез, уреаплазмоз.
Медики предполагают, что возбудители синдрома Рейтера, имеющие антигенное строение, способствуют возникновению определенных реакций иммунной системы у людей с генетической предрасположенностью.
При инфицировании хламидиозом в органах мочеполовой системы (уретре, простате или матке) появляется воспаление, из этого очага патогенные бактерии с током крови разносятся в ткани, в том числе и суставные, способствуя развитию аутоиммунной аллергии.
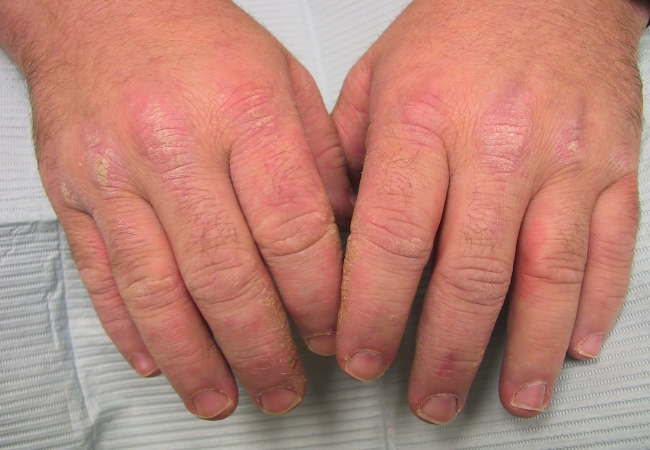
Разновидности заболевания
Болезнь Рейтера у мужчин и женщин имеет две стадии протекания:
- инфекционную, при которой хламидии попадают в уретру;
- иммунопатологическую, когда развивается патология суставов и конъюнктивы.
Также существуют две формы патологии:
- Спорадическая, при которой заболеванию предшествует венерическая болезнь.
- Постэнтероколитическая форма, когда патологии предшествует энтероколит, вызванный уреаплазмой, сальмонеллой, дизентерией, шигеллой или иерсинией.
Заболевание может протекать от шести месяцев (острая стадия) до одного года (затяжная стадия), затем оно приобретает хроническую форму.
Эпидемиология
Болезнь Рейтера является самой частой патологией суставов воспалительного характера у мужчин молодого возраста, реже заболевание поражает женщин и детей. Последние обычно заражаются во время рождения от инфицированной матери.
В 90% случаев патология обнаруживалась у половых партнеров инфицированных людей. Данное заболевание может передаваться по наследству. Обычно синдром Рейтера развивается у тех, кто является носителем антигена HLA В27. По статистическим данным заболевание проявляется у мужчин и женщин в соотношении 10:1.
Причины развития недуга
Чаще всего болезнь развивается из-за бактерий Chlamydia trachomatis, которые при воздействии на организм неблагоприятных факторов трансформируются в L-форму, что способны паразитировать внутри здоровых клеток длительный период времени. Это способствует развитию хронической формы заболевания, которая провоцирует появление у мужчин уретритов, а у женщин цервицитов, циститов или сальпингитов. Женщины, что имеют данные патологии и являются носителями хламидийной инфекции, редко болеют артритами урогенного характера.
С кровотоком хламидии разносятся по организму, оседая в органах и тканях, поэтому заболевание имеет несколько очагов поражения. Инфекция передается половым и бытовым путем. Ее можно обнаружить на слизистой оболочке уретры, цитоплазме клеток синовии и конъюнктивы. В некоторых случаях болезнь Рейтера (фото предоставляется в статье) провоцируется сальмонеллами, шигеллами и появляется после энтероколита. В 95% случаев патология передается по наследству.
Симптомы и признаки патологии
Болезнь Рейтера симптомы сразу все не проявляет. У 40% людей первые признаки заболевания проявляются через три месяца после инфицирования. Сначала происходит поражение органов мочеполовой системы, симптоматика при этом неярко выражена. Затем развивается артрит и конъюнктивит. Обычно люди сначала приходят к терапевту и окулисту, которые проводят лечение, не дающее результатов из-за того, что не берется во внимание урогенитальная инфекция.
Обычно патология проявляется триадой признаков: заболевание органов мочеполовой системы, артрит и конъюнктивит. В некоторых случаях поражаются слизистые оболочки ротовой полости, в результате чего развивается стоматит или глоссит, и полового члена, провоцируя появление баланита или баланопостита.
Также патология поражает кожный покров в 50% случаев. На нем образуются папулы красного цвета, участки гиперемии с шелушением и трещинами. Обычно такие явления наблюдаются на коже ладоней и стоп. Нередко в патологический процесс включаются нервная и сердечно-сосудистая системы. Возможно развитие пневмонии, нефритов, плеврита, миокардита и прочих недугов.
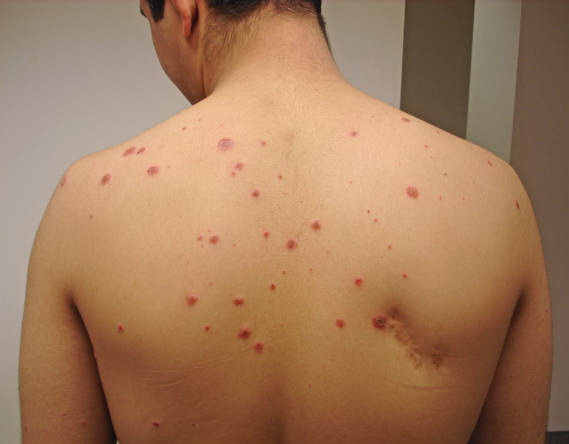
При затяжном протекании заболевания возможно увеличение температуры тела до высоких показателей, наблюдается интоксикация организма, развивается анемия, атрофия мышц, снижение массы тела. При сложной форме патологии происходит нарушение функциональности суставов, расстройство зрения, эрекции, развивается бесплодие, увеличиваются лимфатические узлы. Позже поражаются почки, сердце и аорта, а также нервная система.
Болезнь Рейтера имеет большое разнообразие симптоматики, которая выражается по-разному. Обычно после первых атак заболевания происходит выздоровление, но иногда оно переходит в хроническую форму. Также патология может рецидивировать, проявляя не все возможные симптомы. Поэтому необходимо своевременно начинать лечение болезни.
Поражение органов мочеполовой системы
Сначала недуг затрагивает мочеполовые органы. Болезнь Рейтера у мужчин симптомы проявляет неярко выраженные. У представителей сильного пола развивается уретрит и простатит, заболевания могут протекать на протяжении нескольких лет. Больные люди испытывают дискомфорт и признаки гиперемии в области уретры, могут наблюдаться скудные выделения слизи, дизурические расстройства, зуд и жжение в мочеиспускательном канале. Высыпания на половых органах обычно диагностируются неправильно, врачи путают их с псориазом.
Болезнь Рейтера симптомы у женщин проявляет в виде цервицита или андексита, что протекают без проявления каких-либо признаков. При таком течении заболевания наличие воспалительного процесса выявляется на основании лабораторных данных. В мазке обнаруживается увеличение количества лейкоцитов.
Поражение органов зрения
Через две недели после уретрита развивается конъюнктивит, в некоторых случаях могут наблюдаться ириты, увеиты, ретиниты или кератиты. Обычно симптоматика проявляется на протяжении семи дней, но бывают и затяжные формы заболевания. Признаки болезни Рейтера могут быть слабовыраженными, в тяжелых случаях возможно снижение остроты зрения или развитие слепоты. Многие медики утверждают, что поражение органов зрения может происходить в результате попадания инфекции через грязные руки из половых органов.
Поражение суставов
Болезнь Рейтера, симптомы и лечение которой будут рассмотрены ниже, проявляется и в виде реактивного артрита, что возникает через один месяц после поражения мочеполовой системы. Суставы ног вовлекаются в патологический процесс асимметрично. Обычно кожный покров в области пораженных суставов гиперемирован, в полостях формируется выпот. На протяжении нескольких дней происходит поражение проксимальных и дистальных суставов, при этом артралгии имеют большую выраженность в утреннее и вечернее время суток.
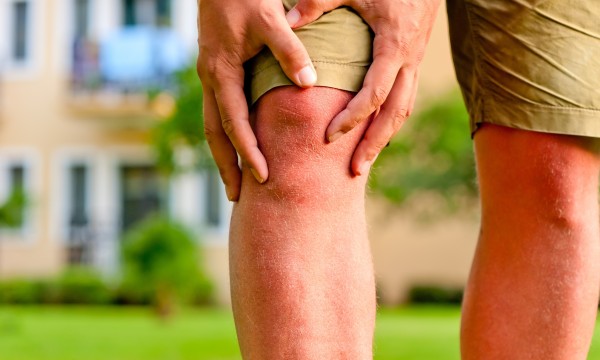
При артрите наблюдаются отечность, деформация пальцев, изменение цвета кожных покровов. Нередко развивается бурсит, пяточные шпоры, тендинит, сакроилеит, которые провоцируют появление болевого синдрома по ночам. Иногда боль может развиваться в области позвоночника, формироваться плоскостопие.
У половины пациентов отмечается полное исчезновение признаков заболевания, у 30% людей артрит со временем рецидивирует, в остальных случаях заболевание протекает в хронической форме, которая характеризуется атрофией мышц и нарушением подвижности суставов.
Диагностика
Во время постановки диагноза симптомы могут проявляться не полностью, так как между поражением трех составляющих проходит некоторый период времени. Поэтому не всегда удается определить связь между поражением мочеполовой системы, суставов и органов зрения.
Сегодня не разработано никаких специфичных анализов для диагностирования патологии. К диагностическим признакам болезни относят острый артрит нижних конечностей, воспаление в мочеполовой системе и органах зрения, поражение эпителия и кожных покровов.
Врач для определения этих признаков назначает лабораторные анализы крови, мочи, исследование мазков и секрета простаты. Проводится цитологическое исследование соскобов эпителия уретры, шейки матки, конъюнктивы и синовиальной жидкости на наличие хламидийной инфекции. В данном случае применяется тест по Романовскому-Гимзе, который имеет чувствительность 95%. Также с этой целью назначается ПЦР, РНГА, ИФА и РСК. Главным признаком патологии выступает обнаружение антигена HLA 27. В качестве инструментальной диагностики применяют рентгенографию суставов.
Врач проводит дифференциацию болезни Рейтера с такими заболеваниями, как ревматоидный и псориатический артриты, анкилозирующий спондилоартрит, гонорейный артрит, ревмокардит, бруцеллезный артрит и так далее.
Терапия болезни
Болезнь Рейтера лечение предполагает комплексное. При этом проходить курс лечения должны оба половых партнера. Врач разрабатывает следующую тактику лечения:
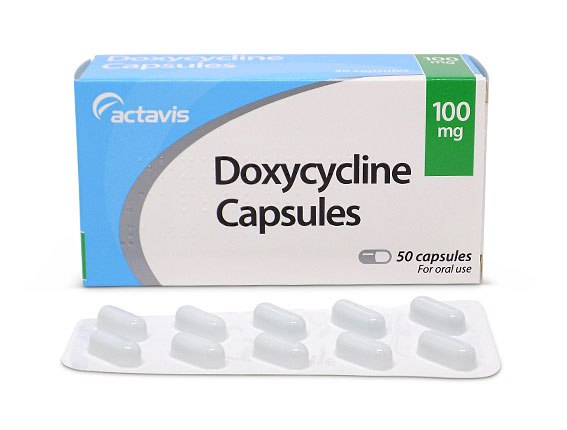
- Терапия антибактериальными препаратами с целью устранения источника инфекции.
- Противовоспалительное лечение суставов.
- Иммунокорекция.
Болезнь Рейтера лечение антибиотиками включает три курса длительностью по три недели каждый. Для этого врач назначает препараты из группы тетрациклинов, макролидов, фторхинолонов. При наличии хронического хламидиоза часто выписывается «Доксициклин». Одновременно с этим необходимо принимать противогрибковые лекарства, витамины, гепатопротекторы и ферменты, иммуномодуляторы, адаптогены. В тяжелых случаях назначается дезинтоксикационная терапия и антигистаминные средства.
Для лечения суставов врач выписывает препараты группы НПВС, нередко проводит пункцию сустава с введением в его полость глюкокортикостероидов. Затем накладывается компресс с раствором обезболивающего препарата и противовоспалительной мази. После устранения симптомов артрита лечение болезни Рейтера у мужчин и женщин предполагает назначение физиотерапии, массажа, родоновых ванн, УВЧ и прочего.
В случае необходимости врач может выписать седативные средства или транквилизаторы. Нередко проводится психотерапия, направленная на выработку у человека установки на выздоровление. При наличии депрессий выписываются антидепрессанты из группы СИОЗ, электросон.

Определение выздоровления
Опытный врач расскажет, какие предполагает болезнь Рейтера симптомы и лечение (фото заболевания приложено выше). При терапии патологии пациент должен находиться под постоянным наблюдением медика. Первая повторная диагностика проводится через один месяц после окончания терапии, вторая – через три месяца. Затем человек должен каждые полгода на протяжении трех лет проходить обследование. Необходимо это для исключения развития рецидивов при хронической форме болезни. Такие пациенты должны находиться под наблюдением уролога, окулиста, дерматовенеролога. Такие меры необходимы для своевременного лечения при появлении угрозы развития поздних осложнений.
Прогноз и профилактика
Прогноз заболевания обычно благоприятный. У половины пациентов недуг через шесть месяцев переходит в ремиссию, но риск развития рецидива остается. У одной четвертой части больных артрит приобретает хроническую форму, провоцируя нарушение функциональности суставов, развитие плоскостопия и атрофии мышц.
Профилактика патологии направлена на предупреждение развития ЗППП, кишечных инфекционных заболеваний, своевременной терапии энтероколитов и уретритов.
Болезнь Рейтера развивается остро, но протекает доброкачественно, периоды обострения чередуются с неожиданными ремиссиями. Важно своевременно пройти обследование для выявления причин развития плохого самочувствия, чтобы иметь возможность пройти эффективное лечение и избавиться от патологии. Врачи рекомендуют вести здоровый и правильный образ жизни, иметь одного постоянного полового партнера.
Синдром Рейтера — Википедия
Материал из Википедии — свободной энциклопедии
Текущая версия страницы пока не проверялась опытными участниками и может значительно отличаться от версии, проверенной 1 марта 2019; проверки требуют 2 правки. Текущая версия страницы пока не проверялась опытными участниками и может значительно отличаться от версии, проверенной 1 марта 2019; проверки требуют 2 правки.Синдром Рейтера (morbus Reiter; Н. Reiter, нем. врач, 1881 — 1969; син.: синдром Рейтера, уретроокулосиновиальный синдром, синдром Фиссенже — Леруа — Рейтера
Заболевание впервые описано в 1916 г. Рейтером и независимо от него Фиссенже (N. A. Fiessinger) и Леруа (Е. A. Leroy) у военнослужащих во время эпидемических вспышек кишечных инфекций.
Развивается, как правило, в молодом возрасте (20—40 лет), преимущественно у мужчин; известны единичные случаи заболевания у детей. Генетическая предрасположенность доказывается наличием у 75—90% больных трансплантационного антигена HLA В27 (в общей популяции встречается лишь в 5—8%)[1].
Часто развиваются кератит и ирит.
Также синдром Рейтера часто вызывается хламидиями и гонококками. Есть 2 формы этого синдрома — спорадическая и эпидемическая.
Инкубационный период 2 недели. Более половины больных являются носителями HLA-B27. Лечение — преимущественно ликвидация возбудителя + НПВС.
Помимо классического проявления синдрома Рейтера возможны изменения кожи (кератодермия, преимущественно на подошвах и ладонях, псориазоподобная сыпь), слизистых оболочек (безболезненный эрозии), поражением суставов (через 1-4 недели после первых симптомов, может сопровождаться лихорадкой, ознобами), возможно развитие подпяточных бурситов, тендовагинита ахиллова сухожилия, изредка развивается атрофация мышц, прилегающих к пораженным суставам, лимфаденит, нарушение сердечного ритма[2].
- ↑ РЕЙТЕРА БОЛЕЗНЬ — Большая Медицинская Энциклопедия (неопр.). xn--90aw5c.xn--c1avg. Дата обращения 16 июля 2019.
- ↑ РЕЙТЕРА БОЛЕЗНЬ — Большая Медицинская Энциклопедия (неопр.). xn--90aw5c.xn--c1avg. Дата обращения 13 августа 2019.
РИХТЕРА СИНДРОМ – это… Что такое РИХТЕРА СИНДРОМ?
- РИХТЕРА СИНДРОМ
M. N. Richter. Generalized reticular cell sarcoma of lymph nodes associated with lymphatic leukemia. [The] American journal of pathology, 1928; 6: 285–299.
Энциклопедический словарь по психологии и педагогике. 2013.
- РИФТ-ВАЛЛИ ЛИХОРАДКА
- РИХТЕРА УЩЕМЛЕНИЕ ГРЫЖИ
Смотреть что такое “РИХТЕРА СИНДРОМ” в других словарях:
Патологическая анатомия гемобластозов — Эта статья предлагается к удалению. Пояснение причин и соответствующее обсуждение вы можете найти на странице Википедия:К удалению/23 августа 2012. Пока процесс обсужден … Википедия
Таруса (город) — Город Таруса Герб … Википедия
Таруса — Город Таруса Герб … Википедия
FACIAUS NERVUS — FACIAUS NERVUS, лицевой нерв, VІI пара черепных нервов, выходит из заднего отдела Варолиева моста. Вместе с п. intermedius Wrisbergi и слуховым нервом он входит во внутреннее слуховое отверстие височной кости. Дальше лицевой нерв отделяется от… … Большая медицинская энциклопедия
Хронический лимфолейкоз
Хронический лимфолейкоз — Википедия
| Хронический лимфолейкоз | |
|---|---|
 Злокачественные клетки в мазке периферической крови | |
| МКБ-10 | C91.191.1 |
| МКБ-10-КМ | C91.1 и C91.10 |
| МКБ-9 | 204.9204.9 |
| МКБ-9-КМ | 204.1[1][2] |
| МКБ-О | 9823/3 |
| OMIM | 109543, 151400, 609630, 612557, 612558, 612559, 151400, 609630, 109543, 612557, 612559 и 612558 |
| DiseasesDB | 2641 |
| MedlinePlus | 000532 |
| eMedicine | med/370 |
| MeSH | D015462 |
Хронический лимфолейкоз, или хронический лимфоцитарный лейкоз (ХЛЛ), — злокачественное клональное лимфопролиферативное заболевание, характеризующееся накоплением атипичных зрелых CD5/CD19/CD23-положительных В-лимфоцитов преимущественно в крови, костном мозге, лимфатических узлах, печени и селезёнке[3].
Хронический лимфолейкоз — одно из наиболее распространённых онкогематологических заболеваний[4]. Также это наиболее частый вариант лейкоза среди представителей европеоидной расы. По непонятным причинам редко встречается среди населения стран Восточной Азии. Дебют заболевания, как правило, происходит в пожилом возрасте — медианный возраст на момент постановки диагноза составляет 70—72 года. Мужчины болеют в 1,5—2 раза чаще, чем женщины. Ежегодная заболеваемость составляет 6,8 случаев на 100 тыс. мужчин и 3,5 случая на 100 тыс. женщин
Предрасположенность к заболеванию передаётся по наследству — риск развития хронического лимфоцитарного лейкоза у непосредственных родственников в 8,5 раз превышает популяционный, однако даже при этом остается ниже 1%[6]. Описаны семейные случаи с относительно высокой пенетрантностью. Большей части случаев ХЛЛ, если не всем, предшествует предлейкозное состояние (моноклональный В-клеточный лимфоцитоз), которое наблюдается у 5—10 % людей в возрасте старше 40 лет и прогрессирует в ХЛЛ с частотой около 1 % в год[7].
Наследственные факторы[править | править код]
Анализ генома людей с наследственным ХЛЛ позволил идентифицировать однонуклеотидные полиморфизмы, ассоциированные с этим состоянием. Полиморфизмы были обнаружены примерно в 30 локусах, например, в генах IRF4, LEF1[en] и BCL2[5].
Факторы окружающей среды[править | править код]
Контакт с агентом «оранж»[8] и инсектицидами[9] повышает риск развития ХЛЛ.
Этиологическая связь ХЛЛ с ионизирующим излучением, вирусными инфекциями, питанием и образом жизни не доказана[5].
Первоначально хронический лимфоцитарный лейкоз рассматривали как онкологическое заболевание, характеризующееся накоплением долгоживущих, но очень редко делящихся иммунологически некомпетентных B-лимфоцитов[10]. Однако исследования с использованием тяжёлой воды показали, что злокачественные клетки пролиферируют, и достаточно быстро — количество новых клеток, образующихся за день, составляет от 0,1 до более чем 1 % от общего числа клеток клона[11]. Причём при высокой скорости пролиферации более вероятно агрессивное течение болезни.
Клеточное микроокружение (ниша) играет большую роль в патогенезе хронического лимфолейкоза. Пролиферация злокачественных клеток происходит в микроанатомических структурах, которые называются пролиферативными центрами, или псевдофолликулами. Псевдофолликулы представляют собой скопления лейкозных клеток, находящихся в контакте со вспомогательными клетками (например, стромальными клетками), которые стимулируют их пролиферацию и выживание[12]. Пролиферативные центры в основном находятся в лимфатических узлах и в меньшей степени в костном мозге[13].
Происхождение злокачественного клона[править | править код]
Злокачественные клетки имеют CD19/CD5/CD23-положительный иммунофенотип и низкий уровень мембранных иммуноглобулинов. Нормальные популяции В-клеток с таким набором поверхностных маркеров неизвестны, что мешает установить, какой тип клеток может давать начало злокачественному клону при ХЛЛ. Анализ транскриптома показал, что опухолевые клетки по набору синтезируемых мРНК похожи на зрелые В-клетки, которые прошли активацию антигеном. В норме таким профилем экспрессии генов обладают В-клетки памяти и В-клетки краевой зоны лимфатических фолликулов, поэтому предполагают, что именно они могут быть предшественниками лейкозных клеток[7].
В отличие от других B-клеточных лейкозов, для ХЛЛ не удалось выявить типичных хромосомных транслокаций, затрагивающих онкогены. Кроме того, крупные хромосомные перестройки редко наблюдаются на ранних стадиях заболевания, так что маловероятно, что они являются первичной причиной ХЛЛ. Однако по мере прогрессирования заболевания такие перестройки происходят: чаще всего это делеции участков хромосом 11, 13 и 17[3].
Характерен абсолютный лимфоцитоз в периферической крови (по данным гемограммы) и костном мозге (по данным миелограммы). На ранних стадиях лимфоцитоз является единственным проявлением заболевания. Пациенты могут предъявлять жалобы на так называемые «конституциональные симптомы» — астению, повышенную потливость, спонтанное снижение массы тела.
Характерна генерализованная лимфаденопатия. Увеличение внутригрудных и внутрибрюшных лимфатических узлов выявляется при ультразвуковом или рентгенологическом обследовании, периферические лимфоузлы доступны пальпации. Лимфатические узлы могут достигать значительных размеров, образовывать мягкие или плотноватые конгломераты. Сдавление внутренних органов не характерно.
На более поздних стадиях заболевания присоединяется гепатомегалия и спленомегалия. Увеличение селезёнки может проявляться ощущением тяжести или дискомфорта в левом подреберье, феноменом раннего насыщения.
За счёт накопления опухолевых клеток в костном мозге и вытеснения нормального гемопоэза на поздних стадиях могут развиваться анемия, тромбоцитопения, редко нейтропения. Поэтому пациенты могут жаловаться на общую слабость, головокружения, петехии, экхимозы, спонтанную кровоточивость.
Анемия и тромбоцитопения также могут иметь аутоиммунный генез.
Для заболевания характерна выраженная иммуносупрессия, затрагивающая преимущественно гуморальный иммунитет (гипогаммаглобулинемия). Из-за этого имеется предрасположенность к инфекциям, например, рецидивирующим простудным заболеваниям и пневмонии.
Необычным клиническим проявлением заболевания может быть гиперреактивность на укусы насекомых.
Для дифференциальной диагностики хронического лимфоцитарного лейкоза с другими лимфопролиферативными заболеваниями необходимо проанализировать количество В-клеток в периферической крови, мазок крови и провести иммунофенотипирование циркулирующих в крови лимфоцитов. Дополнительно для определения прогноза (но не схемы лечения) иногда проводят цитогенетическое исследование, определяют мутационный статус локуса IgVH, количество ZAP-70 или CD38 в клетках ХЛЛ, количество CD23, тимидинкиназы и β2-микроглобулина в сыворотке крови, а также анализируют биоптат или аспират костного мозга[14].
Анализ крови[править | править код]
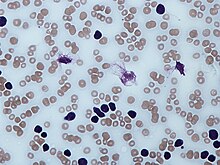 Тени Гумпрехта в мазке крови
Тени Гумпрехта в мазке кровиНеобходимым критерием диагноза хронического лимфоцитарного лейкоза является повышение абсолютного числа В-лимфоцитов в крови до или более 5×109/л. Кроме того, эти лимфоциты должны иметь характерный иммунофенотип: на их поверхности должны обнаруживаться CD19, CD5, CD23, небольшие количества CD20 и CD79b, а также лёгкие цепи иммуноглобулинов[15].
В мазке крови обнаруживаются опухолевые клетки, которые имеют морфологию зрелых (малых) лимфоцитов: «штампованное» ядро с конденсированным хроматином без ядрышка, узкий ободок цитоплазмы. Характерно наличие так называемых теней Гумпрехта, которые представляют собой лейкозные клетки, разрушившиеся в процессе приготовления мазка. Помимо малых лимфоцитов в мазке могут присутствовать более крупные или атипичные клетки, иногда отмечается существенная (более 10 %) примесь омоложенных клеток (пролимфоцитов и параиммунобластов), требующая проведения дифференциального диагноза с пролимфоцитарным лейкозом[14].
Иммунофенотипирование[править | править код]
Иммунофенотипирование лимфоцитов методом проточной цитометрии обязательно для подтверждения диагноза. Высокочувствительная проточная цитометрия позволяет обнаруживать одну злокачественную клетку на 10 000 нормальных лейкоцитов[15]. В качестве диагностического материала обычно используется периферическая кровь. Для клеток ХЛЛ характерен аберрантный иммунофенотип: одновременная экспрессия (коэкспрессия) Т-клеточного маркера CD5 и В-клеточных маркеров CD19 и CD23[14]. Количество В-клеточных маркеров CD20, CD79b и мембраносвязанных иммуноглобулинов IgM и IgD понижено по сравнению с нормальными В-клетками[3]. В дополнение к этому выявляется клональность. Диагноз ХЛЛ также может быть установлен на основании данных иммуногистохимического исследования биоптата лимфатического узла или селезёнки.
Подозрение на хронический лимфоцитарный лейкоз также возникает в случае обнаружения у в остальном здоровых людей увеличения абсолютного числа клональных B-лимфоцитов соответствующего иммунофенотипа, даже если общее их количество в периферической крови меньше 5000/микролитр. Если этому признаку не сопутствует лимфаденопатия или органомегалия, цитопении или другие признаки заболевания, такое состояние диагностируется как моноклональный B-лимфоцитоз[14]. Согласно исследованию, проведённому на 1520 участниках в возрасте от 62 до 80 лет с нормальными показателями крови, моноклональный B-лимфоцитоз с иммунофенотипом ХЛЛ обнаруживается у 5 % людей в этой возрастной группе. Такой лимфоцитоз может прогрессировать в ХЛЛ со скоростью около 1 % в год[15].
Цитогенетическое исследование[править | править код]
Цитогенетическое исследование проводится методом стандартного кариотипирования или FISH. Задача исследования — выявление хромосомных мутаций, часть из которых имеет прогностическую значимость. Из-за возможности клональной эволюции исследование должно повторяться перед каждой линией терапии и в случае возникновения рефрактерности.
Стандартное кариотипирование возможно только для клеток в метафазе клеточного цикла. Так как злокачественные клетки при ХЛЛ обладают низкой митотической активностью, для получения необходимого для анализа количества метафаз требуется применение митогенов. Но даже в таком случае хромосомные мутации удаётся обнаружить только в 40—50 % случаев[16].
Интерфазная FISH при хроническом лимфоцитарном лейкозе не требует применения митогенов и отличается большей чувствительностью. При анализе используют локус-специфичные зонды, позволяющие выявлять наиболее распространённые хромосомные перестройки (как правило делеции). Этот метод позволяет детектировать хромосомные мутации в более чем 80 % случаев хронического лимфоцитарного лейкоза[16].
У каждого отдельного пациента может быть обнаружена одна, две и более стандартных мутации. Исследование, проведённое на 325 пациентах с хроническим лимфоцитарным лейкозом, позволило установить иерархию кариопитов по их прогностической способности: del17p, del11q, трисомия 12, нормальный кариотип и del13q. Если у пациента обнаружено больше одной мутации, то прогноз делают по той из них, которая находится выше в этом списке[16].
Результат FISH-исследования. Определяется только один аллель гена ATM (зеленая метка). У пациента имеется del11q22.3.Хромосомные перестройки ассоциированы с определёнными клиническими характеристиками заболевания[16]:
- del13q14 выявляется в ~55 % случаев, делеция может быть моно- и биаллельной, заболевание, как правило, диагностируется на ранней стадии и развивается медленно, прогноз благоприятный;
- трисомия по хромосоме 12 выявляется в ~15 % случаев, прогноз обычный;
- del11q выявляется в ~15 % случаев, болезнь диагностируют на более поздних стадиях, выше вероятность проявления конституциональных симптомов, болезнь быстро прогрессирует, прогноз неблагоприятный, данная мутация может ассоциироваться с резистентностью к алкилирующим химиопрепаратам;
- del17p13 выявляется в ~7 % случаев, болезнь диагностируют на более поздних стадиях, выше вероятность проявления конституциональных симптомов, болезнь быстро прогрессирует, прогноз наиболее неблагоприятный, клоны часто бывают устойчивы к стандартным схемам химиотерапии с использованием алкилирующих препаратов и/или аналогов пурина[14];
- del6q21 характеризуется неблагоприятным прогнозом[3].
Другие методы[править | править код]
Рутинный физикальный осмотр позволяет получить достаточное представление о клинической динамике, поскольку заболевание носит системный характер. Выполнение УЗИ и компьютерной томографии для оценки объёма внутренних лимфоузлов не является обязательным вне клинических исследований.
Тест на гемолитическую анемию из-за высокой частоты аутоиммунных осложнений при ХЛЛ необходим даже при отсутствии её явных клинических проявлений. Рекомендуется проводить прямую пробу Кумбса, подсчёт числа ретикулоцитов и определение уровня фракций билирубина.
Как правило проведение биопсии костного мозга не требуется для поставки диагноза. Анализ биоптата может помочь сделать прогноз относительно скорости развития болезни, но последние наблюдения показывают, что использование других прогностических маркеров может давать лучшие результаты. Однако анализ аспирата или пунктата костного мозга может понадобиться для выяснения причин цитопении (специфическое поражение костного мозга или аутоиммунное осложнение) путём исследования миелограммы[14].
Некоторые дополнительные тесты используются для предсказания скорости прогрессирования заболевания, но не влияют на выбор схемы лечения. К таким тестам относится определение наличия соматических мутаций в вариабельной области генов тяжёлых цепей иммуноглобулинов (IgVH) и определение количества CD38 и ZAP-70 в клетках, поражённых хроническим лимфоцитарным лейкозом. IgVH без мутаций указывают для более агрессивную болезнь и менее благоприятный прогноз[17][18][19]. Экспрессия CD38 и ZAP-70 коррелирует с отсутствием мутаций в IgVH и плохим прогнозом. Однако пока не до конца ясно, являются ли эти молекулы независимыми прогностическими факторами[14]. Также на агрессивность болезни указывают повышенное количество тимидинкиназы[20], CD23[21] и β2-микроглобулина[22] в сыворотке крови.
Используются системы стадирования, предложенные K. Rai[23] и J. Binet[24]. Оригинальная система Rai была модифицирована с целью снизить количество определяемых групп риска с 5 до 3[14]. Обе системы опираются на данные физического осмотра и стандартные лабораторные анализы и легки в применении. В них отражено естественное течение заболевания — постепенное накопление опухолевой массы. Стадирование позволяет делать прогнозы о выживаемости: прогноз пациентов на поздних стадиях может быть хуже, чем на более ранних. Однако эти системы не дают возможности прогнозировать индивидуальный риск прогрессирования заболевания и выживания на ранних стадиях (стадии 0-II по Rai, A по Binet)[16]. По этой причине стали широко использовать другие прогностические маркеры, такие как цитогенетические характеристики злокачественных клонов, мутационный статус локуса IgVH и количество ZAP-70 или CD38.
|
| |||||||||||||||||||||||
Хронический лимфолейкоз является практически неизлечимым медленнопрогрессирующим (индолентным) заболеванием.
Лечение не начинается сразу после подтверждения диагноза. Заболевание может сохранять стабильность годами, иногда в течение всей жизни больного. Часто наблюдается волнообразное течение с периодами увеличения и уменьшения опухолевого объема. Решение о необходимости начала терапии принимается обычно после периода более или менее длительного наблюдения.
Показания для начала лечения сформулированы в современных рекомендациях. Они отражают картину активной прогрессии заболевания, приводящей к ухудшению медицинского состояния больного и/или качества его жизни.
Из-за системного характера заболевания радиотерапия при хроническом лимфоцитарном лейкозе не применяется. Стандартом терапии являются химиотерапевтические режимы с включением нуклеотидных аналогов, алкилирующих препаратов и моноклональных антител.
Один из наиболее эффективных режимов — «FCR» (англ. fludarabine, cyclophosphamide, rituximab). Он позволяет получить полную ремиссию примерно у 85 % больных низкой группы риска. Однако этот режим имеет побочные эффекты, которые не позволяют использовать его для пациентов пожилого возраста. Кроме того, режим малоэффективен для больных группы высокого риска (например, имеющих делецию 17p)[25].
Активно исследуется возможность применения в терапии алкилирующего препарата бендамустин.
Резистентость к цитостатикам, как правило, обусловлена нарушением механизмов инициации апоптоза в ответ на повреждения ДНК в клетках опухоли. Наиболее типичны мутации гена TP53, приводящие к его инактивации. Клетки с инактивированным p53 не погибают при накоплении повреждений генома. Более того, мутации, индуцированные цитостатиками, могут давать таким клеткам дополнительное преимущество за счет активации онкогенов или инактивации антионкогенов. Таким образом, мутагенез, индуцированный цитостатиками, может являться двигателем клональной эволюции.
У пациентов с резистентным течением в настоящее время используются высокие дозы глюкокортикостероидов, алемтузумаб (моноклональное антитело к CD52[26]), режимы, его содержащие, а также аллогенная трансплантация костного мозга.
Проведение интенсивной химиотерапии и трансплантации костного мозга у пожилых может быть затруднено плохим соматическим статусом и наличием серьезных сопутствующих заболеваний. В этой группе больных часто используется хлорамбуцил или комбинации на его основе.
Новые препараты (леналидомид, BGB-3111, акалабрутиниб, дувелисиб, умбралисиб) и комбинированные режимы на их основе в настоящее время проходят заключительные этапы клинических испытаний.
Существует также значительное количество новых экспериментальных подходов к терапии хронического лимфоцитарного лейкоза, эффективность и безопасность которых окончательно не установлена.
В последние годы показана высокая эффективность ингибиторов тирозинкиназ Btk[27][28] (ибрутиниб, акалабрутиниб и др.) и PI3Kdelta (иделалисиб и др.), а также высокоселективного ингибитора Bcl-2 (венетоклакс). В 2014 году FDA (Управление по санитарному надзору за качеством пищевых продуктов и медикаментов США) выдало разрешение на применение ибрутиниба у больных ХЛЛ, предварительно прошедших как минимум один курс лечения[29]. Данные таргетные препараты обладают высокой активностью даже у пациентов с неблагоприятным прогнозом (del17p) и относительно малотоксичны. В то же время, недостатком является их крайне высокая стоимость.
По клиническим проявлениям хронический лимфоцитарный лейкоз является довольно гетерогенным заболеванием: болезнь может протекать длительно без прогрессии или, наоборот, очень агрессивно[7]. Примерно в 30 % случаев болезнь прогрессирует медленно, так что смерть наступает по причине, не связанной с болезнью. В 15 % случаев смерть от болезни и/или побочных эффектов лечения наступает в течение 2—3 лет с момента постановки диагноза. В остальных случаях болезнь медленно прогрессирует в течение 5—10 лет, после чего наступает терминальная стадия заболевания, за которой следует смерть[30]. В случае пациентов из группы низкого риска медиана выживаемости от момента постановки диагноза достигает 8—10 лет. Известен ряд факторов, которые позволяют прогнозировать результаты лечения и продолжительность жизни, в том числе:
- Наличие или отсутствие признаков соматической гипермутации в генах вариабельных фрагментов иммуноглобулинов В-клеточного рецептора,
- Использование определенных V-генов в структуре В-клеточного рецептора (например, VH3—21),
- Уровень экспрессии тирозинкиназы Zap-70,
- Уровень экспрессии поверхностного маркера CD38,
- Хромосомные мутации del17p, del11q, затрагивающие гены TP53 и ATM,
- Уровень бета-2-микроглобулина в сыворотке крови,
- Стадия заболевания по Rai и Binet,
- Время удвоения числа лимфоцитов периферической крови и т. д.
Опухолевая трансформация, при которой клетки клона приобретают новые характеристики, делающие их похожими на диффузную крупноклеточную лимфому, носит название синдром Рихтера. Прогноз при наличии трансформации крайне неблагоприятный.
- ↑ Disease Ontology release 2019-05-13 — 2019-05-13 — 2019.
- ↑ Monarch Disease Ontology release 2018-06-29sonu — 2018-06-29 — 2018.
- ↑ 1 2 3 4 Chiorazzi N., Rai K. R., Ferrarini M. Chronic lymphocytic leukemia // N Engl J Med. — 2005. — Т. 352, вып. 8. — С. 804—815. — PMID 15728813.
- ↑ Jemal A., Siegel R., Xu J., Ward E. Cancer statistics, 2010 // CA Cancer J Clin. — 2010. — Т. 60, вып. 5. — С. 277—300. — DOI:10.3322/caac.20073. — PMID 20610543.
- ↑ 1 2 3 Kipps T. J., Stevenson F. K., Wu C. J., Croce C. M., Packham G., Wierda W. G., O’Brien S., Gribben J., Rai K. Chronic lymphocytic leukaemia. (англ.) // Nature reviews. Disease primers. — 2017. — Vol. 3. — P. 16096. — DOI:10.1038/nrdp.2016.96. — PMID 28102226. [исправить]
- ↑ Cerhan J. R., Slager S. L. Familial predisposition and genetic risk factors for lymphoma. (англ.) // Blood. — 2015. — Vol. 126, no. 20. — P. 2265—2273. — DOI:10.1182/blood-2015-04-537498. — PMID 26405224. [исправить]
- ↑ 1 2 3 Gaidano G., Foà R., Dalla-Favera R. Molecular pathogenesis of chronic lymphocytic leukemia // J Clin Invest. — 2012. — Т. 122, вып. 10. — С. 3432-3438. — DOI:10.1172/JCI64101. — PMID 23023714.
- ↑ Baumann Kreuziger L. M., Tarchand G., Morrison V. A. The impact of Agent Orange exposure on presentation and prognosis of patients with chronic lymphocytic leukemia. (англ.) // Leukemia & lymphoma. — 2014. — Vol. 55, no. 1. — P. 63—66. — DOI:10.3109/10428194.2013.794267. — PMID 23573826. [исправить]
- ↑ Schinasi L. H., De Roos A. J., Ray R. M., Edlefsen K. L., Parks C. G., Howard B. V., Meliker J. R., Bonner M. R., Wallace R. B., LaCroix A. Z. Insecticide exposure and farm history in relation to risk of lymphomas and leukemias in the Women’s Health Initiative observational study cohort. (англ.) // Annals of epidemiology. — 2015. — Vol. 25, no. 11. — P. 803—810. — DOI:10.1016/j.annepidem.2015.08.002. — PMID 26365305. [исправить]
- ↑ Dameshek W. Chronic lymphocytic leukemia — an accumulative disease of immunologically incompetent lymphocytes // Blood. — 1967. — Т. 29, вып. 4:Suppl. — С. 566—584. — PMID 6022294.
- ↑ Messmer B. T., Messmer D., Allen S. L., Kolitz J. E., Kudalkar P., Cesar D., Murphy E. J., Koduru P., Ferrarini M., Zupo S., Cutrona G., Damle R. N., Wasil T., Rai K. R., Hellerstein M. K., Chiorazzi N. In vivo measurements document the dynamic cellular kinetics of chronic lymphocytic leukemia B cells // J Clin Invest. — 2005. — Т. 115, вып. 3. — С. 755—764. — DOI:10.1172/jci23409.
- ↑ D’Cruz O. J., Uckun F. M. Novel Bruton’s tyrosine kinase inhibitors currently in development // Onco Targets Ther. — 2013. — Т. 6. — С. 161—176. — DOI:10.2147/OTT.S33732. — PMID 23493945.
- ↑ Krysov S., Dias S., Paterson A., Mockridge C. I., Potter K. N., Smith K. A., Ashton-Key M., Stevenson F. K., Packham G. Surface IgM stimulation induces MEK1/2-dependent MYC expression in chronic lymphocytic leukemia cells. (англ.) // Blood. — 2012. — Vol. 119, no. 1. — P. 170—179. — DOI:10.1182/blood-2011-07-370403. — PMID 22086413. [исправить]
- ↑ 1 2 3 4 5 6 7 8 Hallek M., Cheson B. D., Catovsky D. et al. Guidelines for the diagnosis and treatment of chronic lymphocytic leukemia: a report from the International Workshop on Chronic Lymphocytic Leukemia updating the National Cancer Institute-Working Group 1996 guidelines // Blood. — 2008. — Т. 111, вып. 12. — С. 5446-5456. — DOI:10.1182/blood-2007-06-093906. — PMID 18216293.
- ↑ 1 2 3 Rawstron A. C., Bennett F. L., O’Connor S. J., Kwok M., Fenton J. A., Plummer M., de Tute R., Owen R. G., Richards S. J., Jack A. S., Hillmen P. Monoclonal B-cell lymphocytosis and chronic lymphocytic leukemia // N Engl J Med. — 2008. — Вып. 359. — № 6. — С. 575-83. — DOI:10.1056/NEJMoa075290. — PMID 18687638.
- ↑ 1 2 3 4 5 Döhner H., Stilgenbauer S., Benner A., Leupolt E., Kröber A., Bullinger L., Döhner K., Bentz M., Lichter P. Genomic aberrations and survival in chronic lymphocytic leukemia // N Engl J Med. — 2000. — Т. 343, № 26. — С. 1910-6. — PMID 11136261.
- ↑ Damle R. N., Wasil T., Fais F., Ghiotto F., Valetto A., Allen S. L., Buchbinder A., Budman D., Dittmar K., Kolitz J., Lichtman S. M., Schulman P., Vinciguerra V. P., Rai K. R., Ferrarini M., Chiorazzi N. Ig V gene mutation status and CD38 expression as novel prognostic indicators in chronic lymphocytic leukemia. (англ.) // Blood. — 1999. — Vol. 94, no. 6. — P. 1840—1847. — PMID 10477712. [исправить]
- ↑ Hamblin T. J., Davis Z., Gardiner A., Oscier D. G., Stevenson F. K. Unmutated Ig V(H) genes are associated with a more aggressive form of chronic lymphocytic leukemia. (англ.) // Blood. — 1999. — Vol. 94, no. 6. — P. 1848—1854. — PMID 10477713. [исправить]
- ↑ Hamblin T. J., Orchard J. A., Ibbotson R. E., Davis Z., Thomas P. W., Stevenson F. K., Oscier D. G. CD38 expression and immunoglobulin variable region mutations are independent prognostic variables in chronic lymphocytic leukemia, but CD38 expression may vary during the course of the disease. (англ.) // Blood. — 2002. — Vol. 99, no. 3. — P. 1023—1029. — PMID 11807008. [исправить]
- ↑ Hallek M., Langenmayer I., Nerl C., Knauf W., Dietzfelbinger H., Adorf D., Ostwald M., Busch R., Kuhn-Hallek I., Thiel E., Emmerich B. Elevated serum thymidine kinase levels identify a subgroup at high risk of disease progression in early, nonsmoldering chronic lymphocytic leukemia. (англ.) // Blood. — 1999. — Vol. 93, no. 5. — P. 1732—1737. — PMID 10029603. [исправить]
- ↑ Knauf W. U., Langenmayer I., Ehlers B., Mohr B., Adorf D., Nerl C. H., Hallek M., Zwingers T. H., Emmerich B., Thiel E. Serum levels of soluble CD23, but not soluble CD25, predict disease progression in early stage B-cell chronic lymphocytic leukemia. (англ.) // Leukemia & lymphoma. — 1997. — Vol. 27, no. 5-6. — P. 523—532. — DOI:10.3109/10428199709058320. — PMID 9477135. [исправить]
- ↑ Gentile M., Cutrona G., Neri A., Molica S., Ferrarini M., Morabito F. Predictive value of beta2-microglobulin (beta2-m) levels in chronic lymphocytic leukemia since Binet A stages. (англ.) // Haematologica. — 2009. — Vol. 94, no. 6. — P. 887—888. — DOI:10.3324/haematol.2009.005561. — PMID 19483161. [исправить]
- ↑ Rai K. R., Sawitsky A., Cronkite E. P., Chanana A. D., Levy R. N., Pasternack B. S. Clinical staging of chronic lymphocytic leukemia // Blood. — 1975. — Т. 46, вып. 2. — С. 219—234. — PMID 1139039.
- ↑ Binet J. L., Auquier A., Dighiero G., Chastang C., Piguet H., Goasguen J., Vaugier G., Potron G., Colona P., Oberling F., Thomas M., Tchernia G., Jacquillat C., Boivin P., Lesty C., Duault M. T., Monconduit M., Belabbes S., Gremy F. A new prognostic classification of chronic lymphocytic leukemia derived from a multivariate survival analysis // Cancer. — 1981. — Т. 48, вып. 1. — С. 198-206. — PMID 7237385.
- ↑ Riches J. C., Ramsay A. G., Gribben J. G. Chronic lymphocytic leukemia: an update on biology and treatment // Curr Oncol Rep. — 2011. — Т. 13, вып. 5. — С. 379-385. — DOI:10.1007/s11912-011-0188-6. — PMID 21773694.
- ↑ Лекарство, применяемое при лейкемии, может стать мощным оружием в борьбе против рассеянного склероза
- ↑ O’Brien S., Furman R. R., Coutre S. E., Sharman J. P., Burger J. A., Blum K. A., Grant B., Richards D. A., Coleman M., Wierda W. G., Jones J. A., Zhao W., Heerema N. A., Johnson A. J., Izumi R., Hamdy A., Chang B. Y., Graef T., Clow F., Buggy J. J., James D. F., Byrd J. C. Ibrutinib as initial therapy for elderly patients with chronic lymphocytic leukaemia or small lymphocytic lymphoma: an open-label, multicentre, phase 1b/2 trial. (англ.) // The lancet oncology. — 2014. — Vol. 15, no. 1. — P. 48—58. — DOI:10.1016/S1470-2045(13)70513-8. — PMID 24332241. [исправить]
- ↑ Byrd J. C., Furman R. R., Coutre S. E., Flinn I. W., Burger J. A., Blum K. A., Grant B., Sharman J. P., Coleman M., Wierda W. G., Jones J. A., Zhao W., Heerema N. A., Johnson A. J., Sukbuntherng J., Chang B. Y., Clow F., Hedrick E., Buggy J. J., James D. F., O’Brien S. Targeting BTK with ibrutinib in relapsed chronic lymphocytic leukemia. (англ.) // The New England journal of medicine. — 2013. — Vol. 369, no. 1. — P. 32—42. — DOI:10.1056/NEJMoa1215637. — PMID 23782158. [исправить]
- ↑ FDA NEWS RELEASE (неопр.). Дата обращения 24 июля 2014.
- ↑ Spaargaren M., de Rooij M. F., Kater A. P., Eldering E. BTK inhibitors in chronic lymphocytic leukemia: a glimpse to the future. (англ.) // Oncogene. — 2015. — Vol. 34, no. 19. — P. 2426—2436. — DOI:10.1038/onc.2014.181. — PMID 24954503. [исправить]
- Патологическая анатомия. Курс лекций. Под ред. В. В. Серова, М. А. Пальцева. — М.: Медицина, 1998
Доктор Рихтер — Википедия
Материал из Википедии — свободной энциклопедии
Текущая версия страницы пока не проверялась опытными участниками и может значительно отличаться от версии, проверенной 27 августа 2019; проверки требуют 7 правок. Текущая версия страницы пока не проверялась опытными участниками и может значительно отличаться от версии, проверенной 27 августа 2019; проверки требуют 7 правок.«Доктор Рихтер» — российский телесериал 2017 года, официальная адаптация американского сериала «Доктор Хаус» / House, M.D.[1][2].
Премьера первого сезона состоялась 13 ноября 2017. Сезон включал в себя 24 серии.
Премьера второго сезона состоялась 19 ноября 2018. Сезон включал в себя 16 серий.
Премьера третьего сезона состоялась 11 ноября 2019. Сезон включал в себя 16 серий.
Большинство эпизодов сериала начинается вне стен городской больницы № 100, где работает врач-диагност Андрей Александрович Рихтер. В начале показываются события, которые предшествуют проявлению симптомов у пациента. На протяжении эпизода команда врачей пытается определить болезнь, вызывающую эти симптомы. Используется метод дифференциальной диагностики, при этом Рихтер руководит обсуждением диагноза. Его интересуют только самые сложные и запутанные случаи — медицинские головоломки. Рутинная работа в больничной поликлинике навевает на него скуку и раздражает. Рихтер избегает разговоров с пациентами, поскольку твердо уверен, что они лгут.
В прошлом Рихтер перенёс сложную травму на бедре (инфаркт четырехглавой мышцы), теперь он ходит, опираясь на трость, и пьёт сильные обезболивающие. Рихтер самоуверен, заносчив, резок и циничен, не обременяет себя соблюдением правил хорошего тона и временами кажется, что сочувствие и сострадание ему чуждо. Строить личные и профессиональные отношения он не умеет и, кажется, не хочет. Люди, находящиеся рядом с ним, становятся объектами насмешек и провокаций, за которыми бывает непросто рассмотреть искреннюю заботу.
- Режиссёры: Андрей Прошкин, Илья Казанков
- Продюсеры: Сергей Мелькумов, Екатерина Ефанова, Александр Роднянский
- Оригинальный сюжет: Дэвид Шор
- Авторы сценария: Александр Родионов, Максим Курочкин, Марина Потапова, Иван Угаров, Варвара Шубина, Вячеслав Дурненков, Полина Бородина при участии Марины Денисевич
- Оператор: Юрий Райский, Антон Костромин
- Композитор: Алексей Айги
- Художник-постановщик: Фёдор Савельев, Вячеслав Чуликов
- Художник по костюмам: Регина Хомская
- Художник по гриму: Ирина Мельникова
- Режиссёр монтажа: Наталья Кучеренко
Телеканал «Россия-1» (ВГТРК) купил лицензию на адаптацию сериала у компании NBCUniversal[3]. Поэтому создатели «Доктора Рихтера», адаптируя сценарий к российской действительности, не могли сильно отходить от концепции оригинала[4].
Реалии работы российской городской клинической больницы № 100, где работает доктор Рихтер вместе с командой, сильно отличаются от повседневной жизни в больницах США. Создатели сериала уверяют, что стремились к максимальной достоверности во всём. В сериале отражены реформы, проходящие в российской медицине, и показаны интриги тех, кто ратует за то или иное решение[5]. Оснащение ординаторской, лаборатории и операционных блоков, даже тележки, каталки, кровати для больных, специфические операционные столы — всё аутентичное[5]. Медицинские компании предоставили для съёмок настоящее оборудование. На съёмках присутствовали действующие врачи, которые консультировали и следили за действиями актёров во время медицинских манипуляций. Актёры перед съёмками осваивали медицинские процедуры в больницах, учили тексты с множеством специальных терминов[5].
Рихтер — циничный, замкнутый и одинокий. С сардоническим чувством юмора, как и Хаус. Но, на мой взгляд, он не лишен сострадания. Для него сострадание выражается через действие, а не через общение или похлопывание по плечу. Мы старались сделать персонажей не пустой калькой, а достоверными для нашей действительности. Герои должны оставаться живыми, чтобы за ними было интересно наблюдать. Поэтому какие-то вещи мы оставляем как в оригинале, но меняем детали и интонацию.
— режиссёр Андрей Прошкин[5]
симптомы и первые признаки, лечение и прогноз
 Болезнь Пика — это редкостная, хроническая и прогрессирующая болезнь ЦНС, характеризующаяся атрофией височных, а также лобных долей коры головного мозга с нарастанием слабоумия.
Болезнь Пика — это редкостная, хроническая и прогрессирующая болезнь ЦНС, характеризующаяся атрофией височных, а также лобных долей коры головного мозга с нарастанием слабоумия.
Заболевание начинается в 50-60 лет, хотя бывают и более поздние или ранние манифестации. Женщины склонны болеть чаще мужчин. А. Пик в 1892 году дал описание случаев сенильной деменции, усиливающихся атрофическим процессом главным образом в височных и лобных долях. Подобные исследования проводили А. Альцгеймер, X. Липман, Е. Альтман.
Высказывания о том, что описанные А. Пиком случаи болезни представляют самостоятельную форму, впервые отметил X. Рихтер. Подтверждением этой нозологической самостоятельности заболевания стали проведенные патологоанатомические исследования, показавшие ряд морфологических особенностей именно этой патологии.
Что это такое?
Болезнь Пика — хроническое и прогрессирующее заболевание центральной нервной системы, встречающееся обычно в возрасте 50 — 60 лет и характеризующееся деструкцией и атрофией коры головного мозга преимущественно в области лобных и височных долей. Средний возраст начала заболевания — 54 года, средняя продолжительность до наступления смерти — 6 лет.
Причины развития
Этиология остается невыясненной. Семейные случаи заболевания наталкивают исследователей на мысль о его наследственном характере. Однако спорадические случаи наблюдаются намного чаще семейных, а в пределах одной семьи чаще болеют братья и сестры, чем родственники в разных поколениях. Среди возможных этиофакторов названы длительные воздействия на головной мозг вредоносных химических веществ.
Сюда же можно отнести применение наркоза, особенно в случаях его частого использования или неадекватного дозирования. Некоторые авторы считают, что болезнь Пика может развиться в связи с перенесенным ранее психическим расстройством. Наряду с этим, исследователи склоняются к мнению, что такие факторы, как интоксикации, черепно-мозговые травмы, инфекции, психические расстройства, гиповитаминоз витаминов гр. В играют лишь провоцирующую роль.
Морфологически определяются атрофические процессы в лобных и височных долях мозга, зачастую больше выраженные в доминантном полушарии. Атрофия затрагивает как кору, так и подкорковые структуры. При этом отсутствуют воспалительные изменения. Сосудистые нарушения не характерны или слабо выражены. Сенильные бляшки и нейрофибриллярные сплетения, типичные для болезни Альцгеймера, отсутствуют. Патогномоничными для болезни Пика считаются внутринейрональные аргентофильные включения.
Классификация
Различают три стадии развития болезни Пика, каждая из которых характеризуется своей клинической картиной и прогнозом. Следует отметить, что развитие недуга на последней стадии является уже необратимым патологическим процессом и часто провоцирует развитие сопутствующих соматических заболеваний.
Выделяют такие стадии развития недуга:
- первая или начальная — наблюдаются негативные изменения в поведении человека. Чаще всего, это эгоистическая ориентация, раздражённость, агрессия к окружающим;
- вторая — прогрессирование клинической картины первой стадии, ухудшаются интеллектуальные способности человека, отсутствует логическое мышление, больной не может сам справляться с элементарными гигиеническими процедурами;
- глубокое слабоумие, человек нуждается в постоянном уходе.
На последней стадии развития заболевания медикаментозная терапия уже не имеет смысла. В этом случае, основой улучшения качества жизни больного является сестринский уход.
Отличие от болезни Альцгеймера
Болезнь Пика и Альцгеймера имеют один общий признак — слабоумие. Дифференцировать одну болезнь от другой можно по таким критериям:
- Возрастные отличия. Болезнь Пика развивается в возрасте пятидесяти лет, в то время как болезнь Альцгеймера практически не диагностируется до шестидесяти лет.
- Память. На первых фазах болезни Пика не отмечается нарушений внимания, человек может ориентироваться на местности, выполнять математический счёт. В случае болезни Альцгеймера основным признаком будет резкое снижение памяти.
- Склонность к агрессии в отношении родственников. Пациенты с нарушением Пика, начиная с ранних этапов заболевания, становятся склонны к бродяжничеству, оказывают сопротивление опеке со стороны близких. У пациентов с болезнью Альцгеймера такие проявления наступают значительно позже.
- Вопрос вседозволенности. При болезни Альцгеймера на первых этапах не возникает личностных изменений, в то время как при болезни Пика выявляется патологическая раскрепощённость, когда больной бездумно подчиняется своим физиологическим инстинктам.
- Речевые нарушения. Первичным симптомом болезни Пика является заметное оскудение словарного запаса. При Альцгеймере на первых этапах ухудшение речи не отмечается, и процесс происходит более медленно.
- Чтение и письмо. Пациенты с заболеванием Альцгеймера практически сразу утрачивают навыки чтения и письма, у людей с болезнью Пика эти умения не утрачивается ещё долго
Симптомы
В начальных стадиях болезни Пика у пациентов наблюдаются такие симптомы: асоциальное поведение, выраженная эгоистичность. Они утрачивают способность контролировать свои поступки, в результате чего становятся эксцентричными – в частности, удовлетворяют базовые инстинкты, в том числе половое влечение, невзирая на окружающих людей и обстановку. Критика к своим поступкам снижена. На этом фоне формируются различные расстройства, например булимия или гиперсексуальность.
На начальных стадиях болезни Пика больные могут пребывать как в состоянии эйфории, так и апатии. У них формируется характерное речевое расстройство, проявляющееся в постоянном повторении одних и тех же анекдотов, фраз, отдельных слов – «симптом граммофонной пластинки». По мере прогрессирования болезни развивается сенсомоторная афазия – теряется способность словесного выражения мыслей, а также понимание речи окружающих. Затем утрачиваются и другие когнитивные функции: счета (акалькулия), письма (аграфия) и чтения (алексия).
В дальнейшем больные теряют способность последовательно выполнять действия (нарушения праксиса), у них изменяется восприятие окружающего мира (агнозия), происходит потеря памяти (амнезия). Исходом становится глубокая деменция. Пациенты не могут обслуживать себя, дезориентированы во времени и пространстве, обездвижены.
Диагностика
При наличии вышеописанной клинической картины следует обратиться психиатру и неврологу. Диагностика при подозрении на болезнь Пика заключается в следующих мероприятиях:
- на основании физикального осмотра больного и личной беседы, врач оценивает психоэмоциональное состояние пациента;
- КТ и МРТ — для оценки состояния головного мозга;
- электроэнцефалография.
Лабораторные анализы, в этом случае, не представляют какой-либо диагностической ценности. В отдельных случаях может понадобиться биохимический анализ крови.
Следует отметить, что данное заболевание необходимо отличать от следующих:
- рак головного мозга;
- болезнь Альцгеймера;
- хорея Гентингтона;
- психические нарушения при атеросклерозе диффузного типа.
На основании полученных результатов обследования, врач может определить степень развития патологического процесса и выбрать наиболее оптимальную тактику поддерживающей терапии.

МРТ
Лечение
На сегодняшний день общей методики для лечения болезни Пика не существует. Всё что может сделать врач — назначить медикаментозное лечение для задержки прогрессирующих изменений и улучшения состояния человека.
В лечении болезни Пика применяют ингибиторы холинэстеразы. Это такие препараты, как Амиридин, Ривастигмин (Экселон), Реминил (Галантамин), Арисепт, а также Глиатилин. Эти лекарства при болезни Пика нормализуют состояние пациентов на раннем этапе заболевания. Хороший эффект имеется от применения длительно (порядка 6 месяцев) блокаторов NMDA (Акатинолмемантин), а также препаратов ноотропного действия (Фенотропил, Аминалон, Ноотропил) и Церебролизин. Купирование продуктивной психотической симптоматики осуществляется мягкими нейролептиками – Терален, Тералиджен, Клопиксол, Хлорпротиксен.
Пациенты с болезнью Пика нуждаются в постоянной психологической поддержке. Больным рекомендовано участие в специальных тренингах, замедляющих прогрессирование заболевания. Прогноз на будущее неблагоприятный. Спустя шесть лет после начала заболевания наступает полное моральное, а также психическое разложение личности, наступает маразм и кахексия. Заболевший для общества становится полностью потерян. Больному нужен обязательный постоянный уход или помещение в специализированную психиатрическую больницу.
Прогноз
Прогноз жизни при деменции, связанной с болезнью Пика неблагоприятный. Болезнь неизлечима, а терапия направлена на ослабление и устранение симптомов. Так как патология развивается постепенно, то ее наличие долгое время остается незаметным. Пациенты поздно обращаются за медицинской помощью. Это приводит к быстрому прогрессированию болезни и ухудшению состояния пациента. Живут пациенты с диагнозом Пика от 6 до 10 лет при хорошем уходе и эффективном лечении.
Часто пациенты с деменцией представляют опасность для самих себя и не способны ухаживать за собой – готовить еду, одеваться, мыться, стирать, убирать. Поэтому таким пациентом необходим постоянный уход и контроль. На третьей стадии патологии пациенты часто становятся обездвиженными вследствие апраксии, дезориентированы. Это приводит к осложнениям – пролежням, инфицированию ран, сепсису, пневмонии. Осложнения часто приводят к гибели пациента.
При хорошем уходе, эффективном медикаментозном и немедикаментозном лечении, окружении пациента заботой и теплом прогрессирование болезни можно замедлить, частично восстановить когнитивные функции головного мозга и значительно улучшить качество жизни пациента с болезнью Пика.